The chemerin-CMKLR1 axis limits thermogenesis by controlling a beige adipocyte/IL-33/type 2 innate immunity circuit

INTRODUCTION
Obesity happens when energy intake chronically exceeds energy expenditure. Adipose tissue is essential in regulating whole-body energy homeostasis and is divided into two major types: white adipose tissue (WAT) and brown adipose tissue (BAT), which are responsible for storage and dissipation of energy, respectively, in response to nutrient and environmental stress (1, 2). Recent studies have revealed that a third type of adipocyte called beige adipocytes can emerge in subcutaneous WAT upon cold exposure in a process known as “beiging” characterized by the expression of uncoupling protein 1 (UCP1), the critical thermogenic effector and browning marker (2, 3). Accumulating evidence has causally linked increased beige fat through genetic manipulation, drugs, or transplantation to enhanced whole-body energy expenditure and improved glucose metabolism (4–6). This is supported by the clinical findings showing the existence of beige-like “brown fat” in adult humans, which correlates to leanness and improved metabolic parameters (7, 8). Thus, understanding the factors that regulate beige fat formation might provide therapeutic targets to enhance energy expenditure and combat obesity.
A variety of environmental cues and mediators have been reported to promote beige fat development, among which cold-induced sympathetic activation is the dominant trigger by releasing norepinephrine (NE) (9). However, because WAT is relatively poorly innervated, beige fat biogenesis is largely dependent on its local environment. In the lean state, type 2 cytokine-associated innate immune cells—including type 2 innate lymphoid cells (ILC2), eosinophils, and alternatively activated M2 macrophages—are enriched in WAT and further elevated upon cold exposure (10). In obesity, a marked shift from type 2 to type 1 cytokine-associated immune cells occurs (11). Thus, type 2 immunity has emerged as a major axis of metabolic regulation by antagonizing obesity-associated inflammatory type 1 immunity and, more recently, by its critical role in facilitating beige fat development. Recent studies highlight an indispensable role of type 2 innate immunity in beige fat biogenesis in response to several physiological stimuli (12–15). Interleukin-33 (IL-33) is a critical upstream cytokine to initiate type 2 immune responses primarily via activation of ILC2 and subsequent secretion of bioactive molecules, which is closely involved in a variety of physiological and pathological conditions (16). Two groups independently reported the important role of the IL-33–ILC2 pathway in functional beige fat biogenesis in subcutaneous WAT by stimulating the proliferation of adipocyte precursors (APs) and beige commitment via IL-4/IL-13 signaling or increasing UCP1 expression via methionine-enkephalin (MetEnk) (17, 18). Further study showed that IL-33 expression during a perinatal age window is sufficient for proper UCP1 expression in thermogenic adipocytes via regulating the splicing of Ucp1 mRNA independent of ILC2 (19). However, in these studies, genetic deletion or pharmacological administration of IL-33 was used, and it remains largely unknown how cold exposure influences IL-33 production in WAT and the underlying regulatory mechanisms.
White adipocytes are considered to link metabolic status to immune responses by secreting adipokines. For example, adiponectin and adipocyte-derived fibroblast growth factor 21 were recently reported to promote cold-induced beige fat via activation of type 2 innate immunity (15, 20). Chemerin is a newly identified adipokine, which is constitutively expressed in abundance by WAT and further increased with obesity in rodents and humans (21). Chemerin was originally known as a chemoattractant in autoimmune diseases by recruiting pDCs or natural killer cells via its functional receptor CMKLR1 (chemerin chemokine-like receptor 1; also named ChemR23) (22, 23). In contrast, we and others reported an anti-inflammatory role of the chemerin-CMKLR1 axis in different types of tissue inflammation via inhibiting inflammatory cytokine levels and myeloid cell infiltration (24–27). Accumulating evidence suggests that chemerin clinically correlates to human obesity (28, 29). However, the exact role of the chemerin-CMKLR1 axis in the regulation of adipobiology and obesity remains unclear. In vitro studies demonstrated some autocrine effects of chemerin on adipocytes that express CMKLR1 at high levels, including the promotion of adipogenesis from 3T3-L1 preadipocytes but not primary preadipocytes and inhibition of inflammatory nuclear factor κB signaling in adipocytes (30–32). Some conflicting in vivo results were also reported about the effects of the chemerin-CMKLR1 axis on adiposity and glucose metabolism by using chemerin overexpression or global ablation of chemerin or CMKLR1 in a diet-induced obese model (32–35). This is likely due to the various effects of CMKLR1 expression in nonadipocytes such as skeletal muscle cells and islet β cells (33, 36). Several studies showed down-regulated gene expression of chemerin in in vitro differentiated beige adipocytes and in WAT after peroxisome proliferator–activated receptor agonist stimulation or cold exposure (37–39). We therefore aimed to investigate how the chemerin-CMKLR1 axis influences adipose tissue homeostasis, in particular, for beige fat formation, and the development of obesity via regulation of adipose-immune communication. In this study, we identify adipose chemerin-CMKLR1 axis as a negative regulator of cold-induced beige fat by interrupting the communication between adipocytes and IL-33–driven type 2 innate immunity, which limits adaptive thermogenesis and facilitates obesity and related metabolic dysfunction.
RESULTS
Cold stimulation specifically reduces the expression of chemerin and CMKLR1 in iWAT
We first confirmed the previous finding (30) that Rarres2 (the gene encoding chemerin) was differentially expressed in different adipose tissue depots with higher levels in WAT including subcutaneous inguinal WAT (iWAT) and epididymal WAT (eWAT) and low levels in BAT of wild-type (WT) mice on C57BL/6 background housed at ambient temperature (22°C) (Fig. 1A). Cold exposure (4°C) for 3 days significantly reduced Rarres2 mRNA levels in iWAT but not in eWAT and BAT as well as liver that is a known primary source of chemerin, particularly under steady state (Fig. 1A). Similar results were obtained by treatment with CL-316,243 (CL; a specific β3-adrenergic agonist) (fig. S1A). Our results suggest a negative correlation between Rarres2 mRNA levels and newly recruited beige adipocytes in iWAT. To directly compare the protein levels of chemerin produced by different types of adipocytes, preadipocytes were enriched from isolated stromal vascular fraction (SVF) of iWAT and BAT and cultured under corresponding adipogenic stimuli. Chemerin was detected at very low levels in the cultures on days 0 and 2 when preadipocytes were committed to the adipocyte lineage but gradually increased over adipocyte differentiation, with much higher levels observed during white adipogenesis than beige adipogenesis (Fig. 1B), suggesting that white adipogenesis favors chemerin expression. In contrast, very low levels of chemerin were detected in the culture of differentiated brown adipocytes (Fig. 1B). We also found a similar expression pattern of Cmklr1 in different adipose tissue depots, and cold stimulation specifically reduced its expression in iWAT (Fig. 1C). In contrast, C-C motif chemokine receptor-like 2 (CCRL2), another receptor that mainly functions to enrich local chemerin concentrations (40), was expressed at very low levels in adipose tissues, which was not affected by cold stimulation (Fig. 1C). Furthermore, flow cytometric analysis revealed that CMKLR1 was highly expressed on mature adipocytes and macrophages but not other leukocytes in iWAT (Fig. 1D). Consistently, Western blotting analysis revealed that CMKLR1 at low levels in primary preadipocytes derived from iWAT was gradually up-regulated over white adipogenesis, confirming that CMKLR1 was highly expressed in mature adipocytes (Fig. 1E). CMKLR1 was expressed at the highest levels in differentiated adipocytes under white adipogenesis and at the lowest levels in differentiated brown adipocytes (Fig. 1F). Collectively, our data demonstrate the predominant expression of chemerin and its receptor CMKLR1 in WAT, which is specifically reduced in iWAT upon cold exposure or β3-adrenergic stimulation.
(A) qPCR analysis of Rarres2 expression in iWAT, eWAT, interscapular BAT, and liver from WT mice housed at 22° or 4°C for 3 days. (B) Chemerin concentrations at different time points in the cultures of preadipocytes from iWAT or BAT that were induced into mature adipocytes under corresponding adipogenic stimuli. (C) qPCR analysis of Cmklr1 and Ccrl2 mRNA expression in different adipose tissue depots from WT mice housed at 22° or 4°C for 3 days. (D) Representative flow cytometric data of CMKLR1 expression in individual cell populations in iWAT from three independent experiments. (E) Representative immunoblot data for CMKLR1 expression at different time points during white adipogenesis. (F) Immunoblot analysis of CMKLR1 expression in adipocytes differentiated from preadipocytes from iWAT or BAT under corresponding adipogenic stimuli and densitometry quantification. Data are represented as means ± SEM. In (A) and (C), n = 6 per group. In (B), dots and error bars represent three replicate wells from a representative of three independent experiments with similar results. In (D) to (F), the representative data from three independent experiments with similar results. *P < 0.05 and ***P < 0.001.
” data-icon-position=”” data-hide-link-title=”0″>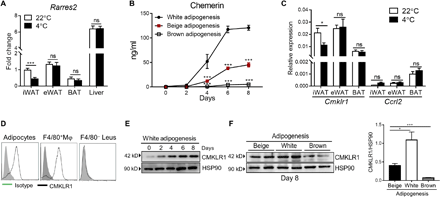
(A) qPCR analysis of Rarres2 expression in iWAT, eWAT, interscapular BAT, and liver from WT mice housed at 22° or 4°C for 3 days. (B) Chemerin concentrations at different time points in the cultures of preadipocytes from iWAT or BAT that were induced into mature adipocytes under corresponding adipogenic stimuli. (C) qPCR analysis of Cmklr1 and Ccrl2 mRNA expression in different adipose tissue depots from WT mice housed at 22° or 4°C for 3 days. (D) Representative flow cytometric data of CMKLR1 expression in individual cell populations in iWAT from three independent experiments. (E) Representative immunoblot data for CMKLR1 expression at different time points during white adipogenesis. (F) Immunoblot analysis of CMKLR1 expression in adipocytes differentiated from preadipocytes from iWAT or BAT under corresponding adipogenic stimuli and densitometry quantification. Data are represented as means ± SEM. In (A) and (C), n = 6 per group. In (B), dots and error bars represent three replicate wells from a representative of three independent experiments with similar results. In (D) to (F), the representative data from three independent experiments with similar results. *P < 0.05 and ***P < 0.001.
Chemerin deficiency promotes cold-induced beige fat biogenesis and thermogenesis
Given that cold is the most potent stimuli for iWAT beiging in vivo, we wondered whether reduced chemerin expression is related to the recruited beige adipocytes in iWAT. We then investigated the role of chemerin in cold-induced iWAT beiging by housing Rarres2−/− mice and WT littermate controls at thermoneutrality (30°C), 22°C, or 4°C for 3 days. There were no differences in body weight and weights of WAT, BAT, and liver between Rarres2−/− mice and WT littermates housed at 22°C or after 3-day cold exposure (fig. S1B). Western blotting analysis revealed obviously higher UCP1 levels in iWAT of Rarres2−/− mice than those of WT littermates upon cold exposure (Fig. 2A). In contrast, comparable UCP1 levels in BAT were observed between Rarres2−/− mice and WT littermates upon cold exposure, whereas UCP1 was undetectable in eWAT under all temperature conditions (Fig. 2A). Histological examination revealed more accumulation of multilocular UCP1-positive adipocytes in iWAT of cold-exposed Rarres2−/− mice (Fig. 2B). Consistently, quantitative polymerase chain reaction (qPCR) analysis revealed that cold exposure caused significantly higher mRNA levels of key thermogenic genes (Ucp1, Ppargc1α, Prdm16, Cidea, Tbx1, Cox8b, and Dio2) but unaltered pan-adipocyte genes (Fabp4 and Adipoq) in iWAT of Rarres2−/− mice (Fig. 2C). In contrast, there were no significant differences in adipocyte morphology, UCP1 expression, or thermogenic gene expression in BAT of Rarres2−/− mice and WT littermates (fig. S1, C and D). Furthermore, Gene Ontology (GO) analysis of RNA sequencing (RNA-seq) profiles revealed marked inductions in catabolic pathways including lipid metabolism pathway, mitochondrial function–related pathway, and brown fat cell differentiation in iWAT of cold-exposed Rarres2−/− mice (Fig. 2D). Chemerin deficiency enhanced cold-induced whole-body energy expenditure, as evidenced by significant increases in core body temperature and oxygen consumption (VO2) in Rarres2−/− mice compared with WT littermates (Fig. 2, E and F). There were no differences in food intake, activity, and respiratory quotient (RQ) between Rarres2−/− mice and WT littermates (fig. S1E). Similarly, CL administration caused significant increases in iWAT beiging and VO2 in Rarres2−/− mice (fig. S2, A to D). NE stimulation that activates all adrenoreceptors and thermogenesis also caused higher VO2 in Rarres2−/− mice than WT mice (Fig. 2G). We also found lower serum chemerin levels in healthy adults with active BAT determined by positron emission tomography–computed tomography (PET-CT) than those without active BAT (Fig. 2H), although such correlation analysis of human participants could not rule out the possibility that lower BMI associated with active BAT may contribute to lower serum chemerin levels. Collectively, these results indicate chemerin as a negative regulator of beige fat biogenesis and thermogenesis.
(A) Immunoblot analysis of UCP1 expression and densitometry quantification in different adipose tissue depots of Rarres2−/− mice and WT littermates housed under different temperature conditions. (B and C) Representative iWAT sections stained with H&E or for UCP1 staining and density quantification (B) and mRNA levels of genes associated with thermogenesis and adipogenesis (C) in iWAT of Rarres2−/− mice and WT littermates (n = 4 to 6). (B) Scale bars, 500 μm (top) and 200 μm (bottom two panels). (D) GO analysis of cold-induced clusters that are expressed at least twofold higher levels in iWAT of Rarres2−/− mice than that of WT littermates (n = 3). (E) Rectal temperatures of Rarres2−/− mice and WT littermates at different time points during a 72-hour cold exposure (n = 4 to 6). (F) VO2 in Rarres2−/− mice and WT littermates housed at 22° or 4°C during one 12-hour light-dark cycle (left) and average VO2 (right) (n = 4 to 6). (G) NE-stimulated VO2 in Rarres2−/− mice and WT littermates (n = 4 to 6). (H) Serum chemerin levels in healthy individuals with active BAT ([18F]FDGneg) and without active BAT ([18F]FDGneg) by ELISA analysis (n = 10). Data are represented as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. Results are representative of three independent experiments.
” data-icon-position=”” data-hide-link-title=”0″>
(A) Immunoblot analysis of UCP1 expression and densitometry quantification in different adipose tissue depots of Rarres2−/− mice and WT littermates housed under different temperature conditions. (B and C) Representative iWAT sections stained with H&E or for UCP1 staining and density quantification (B) and mRNA levels of genes associated with thermogenesis and adipogenesis (C) in iWAT of Rarres2−/− mice and WT littermates (n = 4 to 6). (B) Scale bars, 500 μm (top) and 200 μm (bottom two panels). (D) GO analysis of cold-induced clusters that are expressed at least twofold higher levels in iWAT of Rarres2−/− mice than that of WT littermates (n = 3). (E) Rectal temperatures of Rarres2−/− mice and WT littermates at different time points during a 72-hour cold exposure (n = 4 to 6). (F) VO2 in Rarres2−/− mice and WT littermates housed at 22° or 4°C during one 12-hour light-dark cycle (left) and average VO2 (right) (n = 4 to 6). (G) NE-stimulated VO2 in Rarres2−/− mice and WT littermates (n = 4 to 6). (H) Serum chemerin levels in healthy individuals with active BAT ([18F]FDGneg) and without active BAT ([18F]FDGneg) by ELISA analysis (n = 10). Data are represented as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. Results are representative of three independent experiments.
Adipocyte-specific deletion of Cmklr1 promotes cold-induced beige fat biogenesis and thermogenesis
The above findings demonstrated that CMKLR1, but not CCRL2, was abundantly expressed in iWAT, and CMKLR1 expression was reduced by cold stimulation. We therefore investigated whether these receptors were involved in cold-induced iWAT beiging. Cmklr1−/− mice, but not Ccrl2−/− mice, recapitulated the phenotype of Rarres2−/− mice with enhanced cold-induced iWAT beiging, as assessed by immunoblotting for UCP1 expression, histological analysis, and qPCR analysis of thermogenic gene expression (Fig. 3, A to C). To exclude the possibility that CMKLR1 or CCRL2 expression in other tissues could influence cold-induced iWAT beiging, we specifically knocked down Cmklr1 or Ccrl2 expression in iWAT by locally injecting adenoviruses expressing specific short hairpin RNA (shRNA) against Cmklr1 (ShCmklr1) or Ccrl2 (ShCcrl2) and control adenoviruses expressing LacZ (fig. S3A). Consistent with the findings in global knockout mice, local knockdown of Cmklr1 but not Ccrl2 significantly reduced cold-induced iWAT beiging in WT mice (fig. S3, B to D), confirming a critical role of adipose CMKLR1 in this model. G protein–coupled receptor 1 (GPR1) was recently identified as the third receptor of chemerin. However, we found that GPR1 was undetectable in iWAT or in vitro differentiated adipocytes at mRNA level, which is in contrast to its relatively abundant expression in eWAT as a previous study reported (41). This suggests that GPR1 expression may be adipose depot–specific or cell type–specific. To further determine the specific cell type in which CMKLR1 expression was required for inhibiting cold-induced iWAT beiging, we generated Cmklr1flox mice (Cmklr1fl/fl) mice (fig. S4, A and B) and specifically deleted CMKLR1 in adipocytes and macrophages, both of which express high levels of CMKLR1 in iWAT, by crossing Cmklr1fl/fl mice with Adipoq-Cre (termed as Cmklr1Adipoq-cre) or Lyz-Cre transgenic mice (Cmklr1Lyz-cre), respectively. The genotypes of both mouse strains were confirmed by undetectable Cmklr1 gene expression in adipocytes or macrophages (figs. S4B and S5A). There were no differences in body weight and weights of WAT, BAT, and liver between Cmklr1Adipoq-cre mice and Cmklr1fl/fl littermates (fig. S4C). Adipocyte-specific deletion of Cmklr1 enhanced cold-induced iWAT beiging, as assessed by immunoblotting for UCP1 expression, histological analysis, and qPCR analysis of thermogenic gene expression (Fig. 3, D to F). In contrast, there were no significant differences in adipocyte morphology, UCP1 expression, or thermogenic gene expression between BAT of Cmklr1Adipoq-cre mice and Cmklr1fl/fl littermates (fig. S4, D and E). Cmklr1Adipoq-cre mice had significantly increased whole-body energy expenditure as assessed by core body temperature and VO2 (Fig. 3, G and H) but similar food intake, activity, and RQ when compared with Cmklr1fl/fl littermates upon cold exposure (fig. S4F). In contrast, macrophage-specific deletion of Cmklr1 had no obvious effects on cold-induced iWAT beiging, as evidenced by comparable cold-induced UCP1 protein levels, accumulation of multilocular UCP1-positive adipocytes, and thermogenic gene expression in iWAT of Cmklr1Lyz-cre mice and Cmklr1fl/fl littermates (fig. S5, B to D), suggesting a dispensable role of CMKLR1 expression in macrophage for cold-induced iWAT beiging. Collectively, these results demonstrate that adipocytic CMKLR1 is required for inhibiting cold-induced beige fat biogenesis and thermogenesis.
(A to C) Immunoblot analysis of UCP1 expression and densitometry quantification (A), H&E staining or immunohistochemical staining for UCP1 and staining density quantification (B), and indicated gene expression (C) in iWAT of WT, Cmklr1−/−, and Ccrl2−/− mice housed at 4°C for 3 days. Scale bars, 200 μm. (D to F) Immunoblot analysis of UCP1 expression and densitometry quantification (D), H&E staining or immunohistochemical staining for UCP1 and staining density quantification (E), and indicated gene expression (F) in iWAT of Cmklr1Adipoq-cre mice and Cmklr1fl/fl littermates. Scale bars, 200 μm. (G) Rectal temperatures of Cmklr1Adipoq-cre mice and Cmklr1fl/fl littermates at different time points during a 72-hour cold exposure. (H) VO2 in Cmklr1Adipoq-cre mice and Cmklr1fl/fl littermates during one 12-hour light-dark cycle (left) and average VO2 (right). Data are represented as means ± SEM. n = 4 to 6. *P < 0.05, **P < 0.01, and ***P < 0.001. Results are representative of three independent experiments.
” data-icon-position=”” data-hide-link-title=”0″>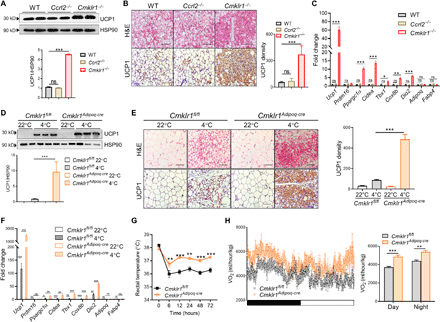
(A to C) Immunoblot analysis of UCP1 expression and densitometry quantification (A), H&E staining or immunohistochemical staining for UCP1 and staining density quantification (B), and indicated gene expression (C) in iWAT of WT, Cmklr1−/−, and Ccrl2−/− mice housed at 4°C for 3 days. Scale bars, 200 μm. (D to F) Immunoblot analysis of UCP1 expression and densitometry quantification (D), H&E staining or immunohistochemical staining for UCP1 and staining density quantification (E), and indicated gene expression (F) in iWAT of Cmklr1Adipoq-cre mice and Cmklr1fl/fl littermates. Scale bars, 200 μm. (G) Rectal temperatures of Cmklr1Adipoq-cre mice and Cmklr1fl/fl littermates at different time points during a 72-hour cold exposure. (H) VO2 in Cmklr1Adipoq-cre mice and Cmklr1fl/fl littermates during one 12-hour light-dark cycle (left) and average VO2 (right). Data are represented as means ± SEM. n = 4 to 6. *P < 0.05, **P < 0.01, and ***P < 0.001. Results are representative of three independent experiments.
Lack of the chemerin-CMKLR1 axis in iWAT promotes cold-induced IL-33 production and type 2 innate immunity
Our group and others previously demonstrated that the chemerin-CMKLR1 axis down-regulates cytokine production at inflammatory sites to inhibit the migration and/or activation of immune cells, including type 2 immune cells (24–26). Given that the lack of the chemerin-CMKLR1 axis promotes cold-induced iWAT beiging but not classical BAT activation, it is likely that the chemerin-CMKLR1 axis modulates the immune environment in iWAT in response to cold stimulation. We found that cold exposure caused increased protein levels of IL-33 and IL-13, which was further enhanced by the lack of chemerin, whereas IL-4 levels were unaltered and IL-5 undetectable in iWAT regardless of cold exposure or chemerin deficiency (Fig. 4A). We then examined the infiltration of immune cells that are reported to respond to IL-33 and found that cold exposure had no effect on the accumulation of CD8+ and CD4+ T cell but mildly increased the numbers of regulatory T cells in iWAT, which, however, was not affected by chemerin deficiency (fig. S6A). Very few, if any, mast cells and basophils were detected in iWAT of mice regardless of cold exposure or chemerin deficiency (fig. S6A). In contrast, cold exposure caused obvious accumulation of ILC2, CD206+ M2-like macrophages, and eosinophils in iWAT of WT mice, which was further enhanced in iWAT of cold-exposed Rarres2−/− mice (Fig. 4, B and C, and fig. S6B). Accordingly, Rarres2−/− mice had higher mRNA levels of M2 macrophage–related genes and proenkephalin A (Penk) (fig. S7A) that was reported to be specifically expressed by ILC2 in iWAT and encodes MetEnk to directly promote UCP1 expression in adipocytes (17). Recent studies showed that IL-33 activates ILC2 to release IL-13, which promotes proliferation and commitment of platelet-derived growth factor receptor α (PDGFRα)+ APs via IL-4Rα to the beige adipocyte lineage (17, 18). Consistent with increased levels of IL-33 and IL-13, higher frequencies and numbers of proliferating 5-bromo-2′-deoxyuridine (BrdU)–positive PDGFRα+ APs were detected in SVF isolated from iWAT of Rarres2−/− mice than those of WT littermates (Fig. 4D and fig. S6C). Moreover, higher mRNA levels of IL-4Rα and beige lineage marker transmembrane protein 26 (TMEM26) and CD137 were detected in PDGFRα+ APs enriched from iWAT SVF of Rarres2− mice (Fig. 4E). Similar to Rarres2−/− mice, higher protein levels of IL-33 and IL-13 and more abundant infiltration of ILC2 and CD206+ M2-like macrophages were detected in iWAT of Cmklr1−/− mice compared with WT littermates (Fig. 4, F and G). In contrast, comparable type 2 immune responses were detected in iWAT of Ccrl2−/− mice and WT littermates (Fig. 4, F and G), which was consistent with unchanged cold-induced beiging in these mice. Accordingly, more proliferating BrdU-positive PDGFRα+ APs with higher mRNA levels of IL-4Rα, TMEM26, and CD137 were detected in PDGFRα+ APs of Cmklr1−/− mice (Fig. 4, H and I). Furthermore, increases in type 2 innate immune responses were recapitulated in iWAT of Cmklr1Adipoq-Cre mice upon cold exposure (Fig. 4, J and K). In addition, there were no differences in mRNA levels of β3-adrenergic receptor (Adrb3) and tyrosine hydroxylase (Th), a rate-limiting enzyme in catecholamine biosynthesis and a marker for sympathetic nerve fibers, between iWAT of Rarres2−/− mice or Cmklr1Adipoq-cre mice and their corresponding littermate controls upon cold exposure (fig. S7, B and C). These data suggest that the chemerin-CMKLR1 axis has no obvious effects on the density of nerve fibers in response to cold stimulation. Collectively, these results demonstrate that the chemerin-CMKLR1 axis in adipocytes is critical for restricting cold-induced IL-33 production and subsequent activation of type 2 innate immunity.
(A to D) ELISA analysis of type 2 cytokine levels in iWAT tissue homogenates (A), representative flow cytometric data of type 2 innate immune cells in total iWAT SVF fraction and proliferating BrdU+PDGFRα+APs (B), absolute numbers of each type 2 innate immune cell (C), and the frequencies of BrdU+PDGFRα+ APs and absolute numbers (D) in iWAT SVF of Rarres2−/− mice and WT littermates. (E) mRNA expression of indicated genes in PDGFRα+ APs enriched from iWAT SVF of Rarres2−/− mice and WT littermates housed at 4°C. (F to H) Type 2 cytokine levels in iWAT tissue homogenates (F), absolute numbers of each type 2 innate immune cell (G), and numbers of BrdU+PDGFRα+ APs in iWAT SVF (H) of WT, Cmklr1−/−, and Ccrl2−/− mice housed at 4°C. (I) mRNA expression of indicated genes in PDGFRα+ APs enriched from iWAT SVF of Cmklr1−/− mice and WT littermates housed at 4°C. (J and K) Type 2 cytokine levels in iWAT tissue homogenates (J) and absolute numbers of each type 2 innate immune cell in iWAT SVF (K) of Cmklr1Adipoq-cre mice and Cmklr1fl/fl littermates. Data are represented as means ± SEM. n = 4 to 6 per group. **P < 0.01 and ***P < 0.001. Results are representative of three independent experiments.
” data-icon-position=”” data-hide-link-title=”0″>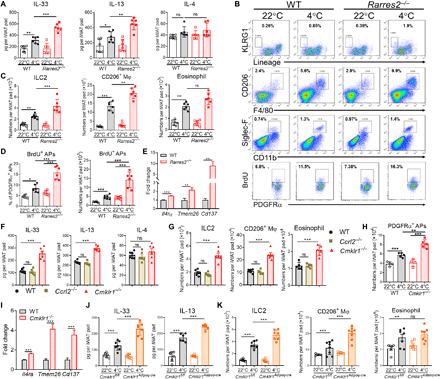
(A to D) ELISA analysis of type 2 cytokine levels in iWAT tissue homogenates (A), representative flow cytometric data of type 2 innate immune cells in total iWAT SVF fraction and proliferating BrdU+PDGFRα+APs (B), absolute numbers of each type 2 innate immune cell (C), and the frequencies of BrdU+PDGFRα+ APs and absolute numbers (D) in iWAT SVF of Rarres2−/− mice and WT littermates. (E) mRNA expression of indicated genes in PDGFRα+ APs enriched from iWAT SVF of Rarres2−/− mice and WT littermates housed at 4°C. (F to H) Type 2 cytokine levels in iWAT tissue homogenates (F), absolute numbers of each type 2 innate immune cell (G), and numbers of BrdU+PDGFRα+ APs in iWAT SVF (H) of WT, Cmklr1−/−, and Ccrl2−/− mice housed at 4°C. (I) mRNA expression of indicated genes in PDGFRα+ APs enriched from iWAT SVF of Cmklr1−/− mice and WT littermates housed at 4°C. (J and K) Type 2 cytokine levels in iWAT tissue homogenates (J) and absolute numbers of each type 2 innate immune cell in iWAT SVF (K) of Cmklr1Adipoq-cre mice and Cmklr1fl/fl littermates. Data are represented as means ± SEM. n = 4 to 6 per group. **P < 0.01 and ***P < 0.001. Results are representative of three independent experiments.
Neutralization of IL-33 or depletion of ILC2 abrogates the enhancement of cold-induced beige fat biogenesis in Rarres2−/− mice
We then asked whether the cold-activated IL-33–ILC2 pathway is required for the enhancement of cold-induced iWAT beiging caused by the absence of the chemerin-CMKLR1 axis. First, IL-33 was locally neutralized by injecting neutralizing IL-33 antibodies or isotype control antibodies into iWAT of Rarres2−/− mice and WT littermates followed by cold exposure. As expected, IL-33 neutralization reduced cold-induced iWAT beiging in WT littermates (Fig. 5, A to C). It completely abrogated the enhancement of cold-induced iWAT beiging in Rarres2−/− mice, as assessed by immunoblotting for UCP1 expression, histological analysis, and qPCR analysis of thermogenic gene expression (Fig. 5, A to C). Moreover, IL-33 neutralization almost completely abrogated the increase of VO2 in cold-exposed Rarres2−/− mice (Fig. 5D). We noted that IL-33 neutralization caused more obvious decreases in UCP1 expression and VO2 in Rarres2−/− mice (Fig. 5, A and D), indicating that cold-induced iWAT beiging and thermogenesis are more dependent on IL-33 when chemerin is absent. IL-33 neutralization also abrogated the increases of type 2 innate immune responses in iWAT of Rarres2−/− mice, as numbers of infiltrating ILC2 and CD206+ M2-like macrophages, as well as IL-13 protein levels and Penk gene expression, were reduced to similar levels observed in similarly treated WT littermates (Fig. 5, E and F), emphasizing IL-33 as an upstream factor of cold-induced type 2 innate immunity. We then examined the contribution of ILC2 to the increased cold-induced iWAT beiging in Rarres2−/− mice. Administration of anti-Thy1.2 antibodies alone reduced cold-induced iWAT beiging in Rarres2−/− mice (fig. S8, A to D). To exclude the potential effect of T cell depletion caused by anti-Thy1.2 antibody treatment, mice were depleted with CD4+ and CD8+T cells followed by administration with anti-Thy1.2 antibodies or isotype controls (fig. S8E). Compared with WT controls with depleted CD4+ and CD8+T cells, similarly treated Rarres2−/− mice still exhibited obvious increases in cold-induced iWAT beiging and mRNA levels of IL-13 and Penk, suggesting that such effects are independent of T cells (Fig. 5, G to J). In contrast, treatment with anti-Thy1.2 antibodies eliminated the differences between WT and Rarres2−/− mice with depleted CD4+ and CD8+T cells (Fig. 5, G to J), suggesting that ILC2 are required for enhanced cold-induced iWAT beiging in Rarres2−/− mice. Collectively, these results demonstrate that the chemerin-CMKLR1 axis restricts the cold-activated IL-33–ILC2 pathway, thereby limiting cold-induced beige fat biogenesis and thermogenesis.
Neutralizing IL-33 antibodies or isotype IgG controls were locally injected into iWAT of Rarres2−/− mice and WT littermates followed by 3-day cold exposure. Immunoblot analysis of UCP1 expression and densitometry quantification (A), H&E staining or immunohistochemical staining for UCP1 and staining density quantification (B), and indicated gene expression (C) in iWAT. Scale bars, 200 μm. (D) Effect of IL-33 neutralization on VO2 in Rarres2−/− mice and WT littermates. (E and F) Numbers of ILC2 and M2 macrophages (E) as well as IL-13 protein levels and Penk gene expression (F) in iWAT. (G to J) WT mice depleted with CD4+ and CD8+ T cells were administered with anti-Thy1.2 antibody or isotype control antibody followed by 3-day cold exposure. Immunoblot analysis of UCP1 expression and densitometry quantification (G), representative iWAT sections stained with H&E or for UCP1 immunohistochemical staining and staining density quantification (H), mRNA levels of indicated genes (I), and IL-13 protein levels and qPCR analysis of Penk mRNA levels (J) in iWAT. Scale bars, 200 μm. Data are represented as means ± SEM. n = 4 to 6. **P < 0.01 and ***P < 0.001. Results are representative of two to three independent experiments.
” data-icon-position=”” data-hide-link-title=”0″>
Neutralizing IL-33 antibodies or isotype IgG controls were locally injected into iWAT of Rarres2−/− mice and WT littermates followed by 3-day cold exposure. Immunoblot analysis of UCP1 expression and densitometry quantification (A), H&E staining or immunohistochemical staining for UCP1 and staining density quantification (B), and indicated gene expression (C) in iWAT. Scale bars, 200 μm. (D) Effect of IL-33 neutralization on VO2 in Rarres2−/− mice and WT littermates. (E and F) Numbers of ILC2 and M2 macrophages (E) as well as IL-13 protein levels and Penk gene expression (F) in iWAT. (G to J) WT mice depleted with CD4+ and CD8+ T cells were administered with anti-Thy1.2 antibody or isotype control antibody followed by 3-day cold exposure. Immunoblot analysis of UCP1 expression and densitometry quantification (G), representative iWAT sections stained with H&E or for UCP1 immunohistochemical staining and staining density quantification (H), mRNA levels of indicated genes (I), and IL-13 protein levels and qPCR analysis of Penk mRNA levels (J) in iWAT. Scale bars, 200 μm. Data are represented as means ± SEM. n = 4 to 6. **P < 0.01 and ***P < 0.001. Results are representative of two to three independent experiments.
Cold-stimulated adipocyte IL-33 production is restricted by the chemerin-CMKLR1 axis via dampening cAMP-PKA signaling
Given the above data showing increased cold-induced IL-33 production in iWAT of Cmklr1Adipoq-cre mice, we reasoned that the action of adipocytic CMKLR1 might inhibit cold-induced IL-33 production via a direct or indirect pathway. Because little is known about how endogenous IL-33 is regulated in iWAT, we first tried to identify the main cellular source for cold-induced IL-33 production. To this end, Il33 mRNA was examined in different types of cells sorted from iWAT of WT or Rarres2−/− mice. We found that CD45+ leukocytes and CD31+ endothelial cells (ECs) expressed very little Il33 mRNA, which was not changed by cold exposure or chemerin deficiency (Fig. 6A). Consistent with recent reports (42, 43), high baseline levels of Il33 mRNA were detected in PDGFRα+ APs from WT mice housed at 22°C, which was only slightly increased by cold exposure or chemerin deficiency (Fig. 6A). These data suggest that PDGFRα+ APs could be the primary source for homeostatic IL-33 but not cold-induced IL-33 production. Despite similarly low Il33 levels in adipocytes from WT and Rarres2−/− mice housed at 22°C, cold exposure markedly up-regulated IL-33 expression in WT adipocytes, with much higher levels in Rarres2−/− adipocytes (Fig. 6A). Furthermore, immunostaining of iWAT tissues revealed that cold exposure caused a higher density of IL-33–positive staining, which was further enhanced by chemerin deficiency. IL-33–positive staining was readily detected in beige-like adipocytes with multilocular lipid droplets determined by perilipin-1 staining (Fig. 6B). Nuclear IL-33 staining was also detected between adipocytes (Fig. 6B), which is consistent with recent studies (42, 43). IL-33 protein expressed by adipocytes was further confirmed by flow analysis of iWAT from cold-exposed Il33fl/fl-eGFP mice (Fig. 7A). These data together indicate adipocytes, most likely newly recruited beige adipocytes, as the main source for cold-induced IL-33 production in iWAT, which is negatively regulated by chemerin. To seek more direct evidence, we measured IL-33 concentrations in the culture of primary preadipocytes under white or beige adipogenic stimuli. IL-33 was detected at baseline levels and gradually increased over beige adipogenesis, reaching the highest levels on day 8 when adipocytes are fully differentiated (Fig. 6C). In contrast, IL-33 levels stayed unchanged over white adipogenesis (Fig. 6C). This is opposite to the pattern of chemerin levels during beige or white adipogenesis (Fig. 1B), suggesting a negative correlation between IL-33 and chemerin expression in adipocytes. Lack of chemerin or CMKLR1 caused significantly higher IL-33 concentrations in the culture of differentiated beige adipocytes (Fig. 6D). We then investigated the molecular mechanism underlying the inhibitory effect of the chemerin-CMKLR1 axis on IL-33 production. Given that CMKLR1 was previously identified as a Gαi protein–coupled receptor to reduce cyclic adenosine 3′,5′-monophosphate (cAMP) (21) and a recent study showing that cAMP agonists enhanced lipopolysaccharide-induced Il33 mRNA in macrophages through a protein kinase A (PKA)–dependent way (44), we speculated that the chemerin-CMKLR1 axis might inhibit cAMP-PKA signaling in adipocytes to restrict IL-33 expression. To test this hypothesis, we measured intracellular levels of cAMP in differentiated adipocytes of different genotypes after beige adipogenic stimuli and found that lack of chemerin or CMKLR1 significantly increased cAMP concentrations (Fig. 6E). Accordingly, markedly increased activation of PKA and cAMP-responsive element-binding protein (CREB) was detected in Rarres2−/− or Cmklr1−/− adipocytes (Fig. 6F). Furthermore, addition of supernatants from culture of WT, but not Rarres2−/−, white adipocytes effectively reduced the increases in IL-33 levels, intracellular levels of cAMP, and activation of PKA and CREB in Rarres2−/− differentiated adipocytes under beige adipogenesis (fig. S9), indicating the bioactivity of chemerin secreted by white adipocytes. To determine whether lack of the chemerin-CMKLR1 axis enhances IL-33 expression via amplifying cAMP-PKA signaling, we stimulated differentiated adipocytes of different genotypes under beige adipogenic stimuli with forskolin, an adenylate cyclase activator to induce cAMP. As shown in Fig. 6D, forskolin treatment caused more IL-33 secretion in the culture of adipocytes of all genotypes, with much higher IL-33 levels in Rarres2−/− or Cmklr1−/− adipocytes than those of WT controls. Moreover, each of the inhibitors for cAMP, PKA, or CREB almost reduced IL-33 secretion in the culture of forskolin-stimulated Cmklr1−/− adipocytes to similar levels observed in those of similarly treated WT adipocytes, emphasizing the importance of cAMP-PKA signaling to induce IL-33 production. Similar change patterns in Il33 mRNA expression were also observed in Cmklr1−/− adipocytes with different treatments (fig. S10). To examine whether such an effect of the chemerin-CMKLR1 axis is conserved in humans, we measured cAMP-PKA signaling and IL-33 production in differentiated human adipocytes by culturing human subcutaneous fat–derived preadipocytes that were transfected with CMKLR1 small interfering RNA (siRNA) or scrambled siRNA under beige adipogenic stimuli. CMKLR1 knockdown caused markedly increased intracellular cAMP levels and activation of PKA and CREB as well as IL-33 production in differentiated human adipocytes (Fig. 6, H to J). Moreover, forskolin treatment caused much more IL-33 secretion by human adipocytes, which was further up-regulated by CMKLR1 knockdown but completely abrogated by treatment with each of the inhibitors for cAMP, PKA, or CREB (Fig. 6, J and K). Collectively, our results demonstrate that the chemerin-CMKLR1 axis inhibits IL-33 production by adipocytes, most likely beige adipocytes, via dampening cAMP-PKA signaling in both rodent and human.
(A) Il33 mRNA expression in indicated cell populations sorted from iWAT (n = 4 to 6). ADs, adipocytes. (B) Representative images of iWAT tissue for IL-33 (red) and Perilipin-1 (PLIN) (green) counterstained with DAPI (blue). Scale bars, 50 μm. Arrowheads indicate IL-33–positive staining in beige-like adipocytes. (C and D) IL-33 concentrations in cultures of iWAT preadipocytes over adipogenesis in response to different stimuli (C) and differentiated adipocytes under beige adipogenesis with or without forskolin treatment. DMSO was used as vehicle control (D). (E and F) cAMP levels (E) and phosphorylation of PKA substrate and CREB (F) in differentiated adipocytes of different genotypes under beige adipogenesis. (G) IL-33 concentrations in cultures of differentiated Cmklr1−/− adipocytes under beige adipogenesis that were pretreated with different inhibitors for cAMP (Rp-8-Br-cAMP), PKA (H-89 2HCL), or CREB (KG-501) followed by forskolin stimulation. (H and I) cAMP levels (H) and immunoblot analysis for CMKLR1 and phosphorylation of PKA substrate and CREB (I) in adipocytes differentiated from human preadipocytes under beige adipogenesis. (J and K) IL-33 concentrations in cultures of differentiated human adipocytes under beige adipogenesis (J) and those treated as described above (K). For the in vitro cell experiments, dots or columns and error bars represent three replicate wells from a representative of three independent experiments with similar results. **P < 0.01 and ***P < 0.001.
” data-icon-position=”” data-hide-link-title=”0″>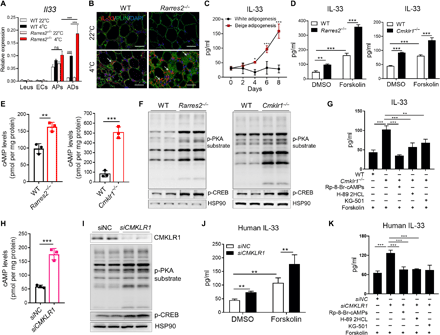
(A) Il33 mRNA expression in indicated cell populations sorted from iWAT (n = 4 to 6). ADs, adipocytes. (B) Representative images of iWAT tissue for IL-33 (red) and Perilipin-1 (PLIN) (green) counterstained with DAPI (blue). Scale bars, 50 μm. Arrowheads indicate IL-33–positive staining in beige-like adipocytes. (C and D) IL-33 concentrations in cultures of iWAT preadipocytes over adipogenesis in response to different stimuli (C) and differentiated adipocytes under beige adipogenesis with or without forskolin treatment. DMSO was used as vehicle control (D). (E and F) cAMP levels (E) and phosphorylation of PKA substrate and CREB (F) in differentiated adipocytes of different genotypes under beige adipogenesis. (G) IL-33 concentrations in cultures of differentiated Cmklr1−/− adipocytes under beige adipogenesis that were pretreated with different inhibitors for cAMP (Rp-8-Br-cAMP), PKA (H-89 2HCL), or CREB (KG-501) followed by forskolin stimulation. (H and I) cAMP levels (H) and immunoblot analysis for CMKLR1 and phosphorylation of PKA substrate and CREB (I) in adipocytes differentiated from human preadipocytes under beige adipogenesis. (J and K) IL-33 concentrations in cultures of differentiated human adipocytes under beige adipogenesis (J) and those treated as described above (K). For the in vitro cell experiments, dots or columns and error bars represent three replicate wells from a representative of three independent experiments with similar results. **P < 0.01 and ***P < 0.001.
(A) The efficiency of IL-33 depletion in adipocytes of cold-exposed mice. (B to E) Immunoblot analysis of UCP1 expression and densitometry quantification (B), H&E staining or immunohistochemical staining for UCP1 and staining density quantification (scale bars, 200 μm) (C), indicated gene expression (D), protein levels of IL-33 and IL-13 and Penk mRNA levels (E), and absolute numbers of each type 2 innate immune cell (F) in iWAT of Il33Adipoq-cre mice and Il33fl/fl littermates housed at 4°C for 3 days. Data are represented as means ± SEM. n = 4. Results are representative of two independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001. MFI, mean fluorescence intensity.
” data-icon-position=”” data-hide-link-title=”0″>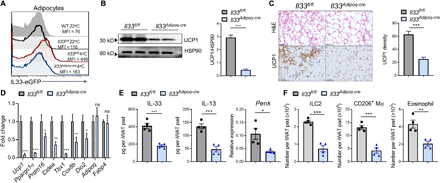
(A) The efficiency of IL-33 depletion in adipocytes of cold-exposed mice. (B to E) Immunoblot analysis of UCP1 expression and densitometry quantification (B), H&E staining or immunohistochemical staining for UCP1 and staining density quantification (scale bars, 200 μm) (C), indicated gene expression (D), protein levels of IL-33 and IL-13 and Penk mRNA levels (E), and absolute numbers of each type 2 innate immune cell (F) in iWAT of Il33Adipoq-cre mice and Il33fl/fl littermates housed at 4°C for 3 days. Data are represented as means ± SEM. n = 4. Results are representative of two independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001. MFI, mean fluorescence intensity.
Adipocyte-specific deletion of Il33 inhibits cold-induced beige fat biogenesis and type 2 innate immunity
To determine whether IL-33 derived from adipocytes is functionally relevant to cold-induced iWAT beiging, we specifically deleted IL-33 in adipocytes by crossing Il33fl/fl mice with Adipoq-Cre (Il33Adipoq-Cre) and used Il33fl/fl littermates as controls. Il33Adipoq-Cre was confirmed by undetectable IL-33 expression in adipocytes upon cold exposure (Fig. 7A). Ablation of IL-33 expression specifically in adipocytes caused a large reduction in the total IL-33 protein level in iWAT upon cold exposure (Fig. 7E). Similar to the phenotype caused by IL-33 neutralization, Il33Adipoq-Cre mice displayed attenuated cold-induced iWAT beiging, as assessed by immunoblotting for UCP1 expression, histological analysis, and qPCR analysis of thermogenic gene expression (Fig. 7, B to D). Accordingly, cold-activated type 2 innate immune responses were attenuated in iWAT of Il33Adipoq-Cre mice compared with those of Il33fl/fl littermates (Fig. 7, E and F). These results demonstrate that cold-induced IL-33 production by adipocytes is sufficient to promote beige fat biogenesis via activation of type 2 innate immunity.
Deletion of adipocytic Cmklr1 protects against diet-induced obesity and glucose intolerance
Increased chemerin levels and defective thermogenic beige fat have been reported to be associated with obesity (28), suggesting a negative correlation. We detected increased chemerin levels, which was primarily derived from adipocytes, in iWAT of WT mice after high-fat diet (HFD) feeding (fig. S11A). We then asked whether the adipose chemerin-CMLKR1 axis had a causal role in HFD-induced obesity. To avoid the influence of CMKLR1 signaling in different cell types on obesity and metabolic parameters, which may cause the previously reported conflicting findings (34, 35), we used Cmklr1Adipoq-cre mice and Cmklr1fl/fl littermates in the HFD-induced obesity model. There was comparable weight gain between Cmklr1Adipoq-cre mice and Cmklr1fl/fl littermates under normal diet (fig. S11B). After chronic HFD challenge, Cmklr1Adipoq-cre mice had less weight gain with significantly reduced fat mass but unchanged lean mass (Fig. 8, A and B). Moreover, adipocyte-specific deletion of Cmklr1 resulted in additional metabolic benefits, including improved glucose intolerance (Fig. 8C), alleviated hepatic steatosis (Fig. 8D), and obvious increases in mRNA levels of type 2 cytokines and thermogenic genes and decreases in inflammatory genes in iWAT (Fig. 8E). However, we noted unimproved insulin sensitivity in Cmklr1Adipoq-cre mice (Fig. 8C). Considering a previous study showing that chemerin promoted insulin resistance in skeletal muscle cells (36), this may be explained by intact CMKLR1 expression in skeletal muscle cells in Cmklr1Adipoq-cre mice. We further assessed energy expenditure in mice after a 3-week HFD when the body weights were similar between the two groups. Cmklr1Adipoq-cre mice exhibited a mild but statistically significant increase in VO2 (Fig. 8F) with no differences in food intake, activity, and RQ when compared with Cmklr1fl/fl littermates (fig. S11C). Collectively, these data demonstrate that Cmklr1 deletion in mature adipocytes increases whole-body energy expenditure and improves HFD-induced obesity and metabolic dysfunction.
(A) Body weight curves of Cmklr1Adipoq-cre mice and Cmklr1fl/fl littermates during HFD feeding. (B to E) Body composition (B), GTT and ITT (C), representative pictures of livers and liver sections stained with H&E (D), and qPCR analysis of type 2 and inflammatory genes and thermogenic genes (E) in iWAT of Cmklr1Adipoq-cre mice and Cmklr1fl/fl mice after 12 weeks on HFD. Scale bars, 500 μm. (F) VO2 in WT and Rarres2−/− mice during one 12-hour light-dark cycle (left) and average VO2 (right) after 3 weeks on HFD. (G) Schematic for the protocol to treat weight-matched obese mice after 12 weeks on normal diet (ND) or HFD by injecting adenoviruses expressing ShCmklr1 or control LacZ vector followed by 1-week cold exposure at 4°C. (H and I) GTT and ITT (H), and qPCR analysis of type 2 and inflammatory genes and thermogenic genes in iWAT (I) of HFD-induced obese mice. (J) Schematic diagram summarizing the mechanism of how the chemerin-CMKLR1 axis inhibits cold-induced IL-33 production and thermogenic beige fat biogenesis. Data are represented as means ± SEM. n = 4 to 6. Results are representative of two independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001.
” data-icon-position=”” data-hide-link-title=”0″>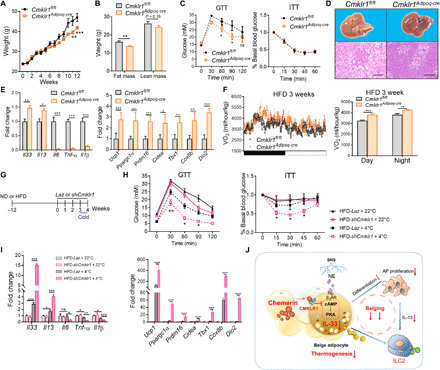
(A) Body weight curves of Cmklr1Adipoq-cre mice and Cmklr1fl/fl littermates during HFD feeding. (B to E) Body composition (B), GTT and ITT (C), representative pictures of livers and liver sections stained with H&E (D), and qPCR analysis of type 2 and inflammatory genes and thermogenic genes (E) in iWAT of Cmklr1Adipoq-cre mice and Cmklr1fl/fl mice after 12 weeks on HFD. Scale bars, 500 μm. (F) VO2 in WT and Rarres2−/− mice during one 12-hour light-dark cycle (left) and average VO2 (right) after 3 weeks on HFD. (G) Schematic for the protocol to treat weight-matched obese mice after 12 weeks on normal diet (ND) or HFD by injecting adenoviruses expressing ShCmklr1 or control LacZ vector followed by 1-week cold exposure at 4°C. (H and I) GTT and ITT (H), and qPCR analysis of type 2 and inflammatory genes and thermogenic genes in iWAT (I) of HFD-induced obese mice. (J) Schematic diagram summarizing the mechanism of how the chemerin-CMKLR1 axis inhibits cold-induced IL-33 production and thermogenic beige fat biogenesis. Data are represented as means ± SEM. n = 4 to 6. Results are representative of two independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001.
Knockdown of adipose Cmklr1 enhances the metabolic benefits of cold stimulation in obese mice
Several clinical studies have demonstrated that cold acclimation recruits new BAT in healthy individuals, which is associated with increased thermogenesis, weight loss, and improved glucose intolerance (6, 45, 46). Given that obesity is associated with a blunted cold-induced adaptive thermogenesis (47), we speculated that interruption of the chemerin-CMKLR1 axis might enhance the metabolic benefits of cold stimulation in obese subjects. To this end, adenoviruses expressing ShCmklr1 or control LacZ vector were locally injected into adiposity-matched WT mice after 12 weeks of HFD feeding followed by 1 week of cold exposure (Fig. 8G and fig. S12A). The efficiency of Cmklr1 knockdown was confirmed by PCR analysis showing very low levels of Cmklr1 mRNA in iWAT of mice that were injected with ShCmklr1-expressing adenoviruses (fig. S12B). Despite unchanged body weight (fig. S12B), cold stimulation caused relatively mildly improvements in glucose intolerance and insulin sensitivity and increases in mRNA levels of IL-33 and IL-13 and thermogenic genes in cold-exposed obese mice that were injected with control LacZ adenoviruses (Fig. 8, H and I), which was paralleled to decreased Cmklr1 mRNA levels in iWAT of these mice (fig. S12A). Local knockdown of Cmklr1 in iWAT greatly enhanced the beneficial effects of cold stimulation on glucose intolerance, insulin sensitivity, and mRNA levels of IL-33 and IL-13 and thermogenic genes in iWAT (Fig. 8, H and I). In addition, CMKLR1 knockdown caused obviously decreased gene expression of proinflammatory cytokines (Fig. 8I). Collectively, these data demonstrate that blockade of adipose CMKLR1 enhances the effects of cold stimulation on type 2 cytokines and thermogenesis and improves glucose metabolism in established obese mice.
DISCUSSION
Given the potential of beige fat to combat obesity and related metabolic disorders, extensive attention has been paid to the factors that promote beige fat biogenesis and adaptive thermogenesis. However, from an evolutional standpoint, energy conservation favors an overall survival advantage of mammals, and therefore, thermogenic beige fat biogenesis needs to be strictly regulated. We here demonstrate that white fat–dominated chemerin-CMKLR1 axis counter-regulates thermogenic beige fat biogenesis to favor energy conservation and contributes to obesity by interrupting adipose-immune communication (Fig. 8J).
Since chemerin was identified as an adipokine one decade ago, its association with adiposity and glucose metabolism has become clear. However, how chemerin influences adipose tissue function remains enigmatic. We here demonstrate that global or adipocyte-specific ablation of chemerin-CMKLR1 axis promoted cold-induced iWAT beiging and adaptive thermogenesis. Moreover, the triggers for iWAT beiging including cold stimulation and β3-adrenergic agonist specifically reduced the expression of chemerin and CMKLR1 in iWAT, emphasizing that the chemerin-CMKLR1 axis serves as a physiological negative regulator of cold-inducing iWAT beiging. Ablation of the chemerin-CMKLR1 axis had no effect on the expression of UCP1 and other thermogenic genes in classical BAT upon cold exposure, suggesting that such inhibitory effects are specific to beige fat formation. CMKLR1 is also reported to be one of the receptors for eicosapentaenoic acid (EPA)–derived lipid mediator resolvin E1 (48). Considering that EPA is directly provided by diet and resolvin E1 is important for suppressing neutrophilic inflammation, white fat–enriched chemerin could be the dominant functional ligand for CMKLR1 in our experimental setting.
Furthermore, we demonstrate that enhanced cold-induced thermogenic beige fat biogenesis caused by ablation of the chemerin-CMKLR1 axis is dependent on elevated IL-33 production and subsequent activation of type 2 innate immune responses in iWAT. Two recent studies showed that IL-33 is constitutively expressed by PDGFRα+ stromal cells in WAT, particularly eWAT, to maintain an anti-inflammatory immune microenvironment via activation of ILC2 or regulatory T cells that protects against obesity (42, 43). Our study here further expands the understanding of the cell source of IL-33 by identifying adipocytes as the main producers of IL-33 in cold-exposed iWAT. Despite very little basal expression in adipocytes, we showed obviously elevated IL-33 expression at both mRNA and protein levels in adipocytes from iWAT of cold-exposed WT mice by qPCR, immunofluorescence staining, and fluorescence-activated cell sorting (FACS) analysis, which was further enhanced in similarly treated Rarres2−/− mice. Given that IL-33 is released as an alarmin in response to cellular damage or stress, our data suggest thermal stress as the stimulus for IL-33 production by adipocytes, most likely those newly recruited beige adipocytes, in iWAT. Using conditional knockout mice, we demonstrated that adipocyte-derived IL-33 is sufficient to promote cold-induced iWAT beiging. Moreover, ILC2 were identified as the major responder to increased IL-33 to mediate cold-induced iWAT beiging in Rarres2−/− mice. This is supported by the evidence that no obvious changes in the accumulation of other IL-33–responding cell types were detected in iWAT and that depletion of ILC2, but not T cells, abrogated the enhancements of cold-induced iWAT beiging and type 2 cytokine levels as well as infiltration of M2 macrophages and eosinophils in Rarres2−/− mice. Together, we propose a previously unrecognized feed-forward circuit between adipocytes and type 2 innate immunity via IL-33, which is critical for thermogenic beige fat formation in vivo, and reveal that the chemerin-CMKLR1 axis negatively regulates such circuit via targeting on adipocytes to restrict IL-33 production, thereby limiting cold-induced iWAT beiging and thermogenesis.
In addition, we demonstrate that the chemerin-CMKLR1 axis controls cold-induced IL-33 production via dampening cAMP-PKA signaling in adipocytes. This could be due to the regulation at transcriptional level, because increased Il33 mRNA levels were detected in Cmklr1−/−adipocytes under beige adipogenic stimuli, which was abrogated by inhibition of cAMP-PKA signaling. However, it is still possible that the chemerin-CMKLR1 axis might also regulate the processing or release of IL-33. The specific molecular mechanism of how the chemerin-CMKLR1 axis regulates IL-33 production by adipocytes awaits further study. Another unsolved issue in our study is whether IL-33 production by adipocytes is beige-selective. We provided some evidence to support such possibility. First, increased IL-33 expression and the appearance of IL-33–positive staining in beige-like adipocytes were observed in iWAT upon cold exposure. Second, in vitro differentiated adipocytes under beige adipogenic stimuli secreted much more IL-33 than those under white adipogenic stimuli. In agreement with our finding, a previous gene profile analysis of forskolin-activated human beige adipocytes showed IL-33 as one of the most up-regulated genes (5). However, considering that both white and beige adipocytes exist in the differentiation culture and cold-exposed iWAT, we cannot rule out the possibility of white adipocytes to produce IL-33. This issue could be clarified in the future when the specific beige marker is identified.
Last, we reveal a causal role of adipocytic CMKLR1 in obesity and related metabolic dysfunction. Despite paradoxical data being reported about HFD-induced obesity in global Cmklr1−/− mice, our study demonstrates that specific deletion of Cmklr1 in adipocyte improved metabolic parameters and increased whole-body energy expenditure after HFD challenge. Moreover, local genetic knockdown of Cmklr1 in iWAT of established obese mice together with a short-term cold stimulation markedly up-regulated the expression of IL-33 and thermogenic genes and further improved glucose metabolism, albeit no obvious change in body weight. These data suggest the likelihood that overactivation of CMKLR1 signaling in adipocytes due to high chemerin levels may be the reason that obese subjects are less sensitive to cold stimulation or β-adrenergic agonists. Thus, simultaneous blockade of adipose chemerin-CMKLR1 axis may be promising to improve the responsiveness to such treatments.
In summary, our study identifies adipose chemerin-CMKLR1 axis as a physiological negative regulator of cold-induced IL-33 and thermogenic beige fat biogenesis and expands the understanding of how adipose-immune communication influences adipose tissue homeostasis in response to environmental stress. Given that the negative regulatory effects of the chemerin-CMKLR1 axis on cAMP-PKA signaling and IL-33 production in adipocytes are conserved in rodents and humans, our study suggests that targeting adipose chemerin-CMKLR1 axis may represent a new strategy to increase beige fat biogenesis and thermogenesis, thereby controlling metabolic diseases.
MATERIALS AND METHODS
Study design
The aim of the study was to determine the role of the chemerin-CMKLR1 axis in the formation of thermogenic beige fat. We performed Western blot, histological analysis, enzyme-linked immunosorbent assay (ELISA), and flow cytometric experiments to evaluate iWAT beiging and type 2 innate immune responses in different knockout mouse strains. The animal experiments were not randomized, and the investigators were not blinded to the allocation of mice to groups during experiments. Experimental replication is indicated in the figure captions.
Experimental animals
Adipoq-Cre, Lyz2-Cre, and Il33fl/fl-eGFP mice were purchased from the Jackson Laboratory. WT mice were purchased from the Chinese Academy of Sciences (Shanghai, China). Rarres2−/− mice and Cmklr1−/− mice were generated by Cyagen Company (Shanghai, China) as previously described (49). Ccrl2−/− mice were generated by Cyagen Company using a TALEN knockout strategy, which resulted in a 1–base pair deletion (T) in exon 2 at approximate position. Ccrl2−/− mice have normal fertility and similar body weight compared with age-matched WT mice during the period that we observed. Cmklr1-floxed (Cmklr1fl/fl) mice were generated by Shanghai Model Organisms Center Inc. (Shanghai, China) by inserting the sequence of flox into exon 3 of murine Cmklr1 gene (MGI#109603). The genotyping primers and the length of PCR production of Rarres2−/−, Cmklr1−/−, Ccrl2−/−, and Cmklr1fl/fl mice are listed in table S1. Mice lacking CMKLR1specifically in adipocytes and macrophages were generated by crossing the Cmklr1fl/fl and Adipoq-Cre lines and Lyz2-Cre line, respectively, and mice lacking IL-33 in adipocytes were generated by mating the Il33fl/fl-eGFP and Adipoq-Cre lines. All transgenic mice are on the C57BL/6J genetic backgrounds, and experiments were conducted with littermate controls. All mice used in experiments were age-matched males between 6 and 8 weeks old and housed under specific pathogen–free conditions. All animal experiments were performed according to protocols approved by the Fudan University Shanghai Medical College.
In vivo studies
For cold challenge experiments, 8-week-old male mice were placed at 4°C in a laboratory incubator for 3 days in groups of three mice per cage. Some mice housed at 30°C for 2 weeks were used as thermoneutral controls. For the HFD-induced obesity study, 6-week-old male mice were fed with either normal diet or HFD diet (60% kcal fat; #D12492, Research Diet). Bodyweight was measured weekly for 12 weeks. Fat mass and lean mass were measured by 1H–nuclear magnetic resonance using an EchoMRI-100 analyzer (EchoMRI LLC). For IL-33 neutralization, polyclonal goat anti-mouse IL-33 antibody (2 μg per mice; #AF3626, R&D Systems) or isotype control antibody was directly administered via bilateral subcutaneous injection into the area adjacent to inguinal fat pads of mice every 2 days starting from 1 day before the 3-day cold exposure. For specific depletion of ILC2, mice that were intraperitoneally injected with anti-CD4 (200 μg per mice; #BE0003-1) and anti-CD8 antibodies (200 μg per mice; #BE0061) 3 days before cold exposure were injected with anti-Thy1.2 antibody (250 μg per mice; #BE0066) 1 day before cold exposure (all from Bio X cell). For CL treatment, mice were intraperitoneally injected with CL (0.5 mg/kg daily) for 5 days.
Human samples
Healthy adults who volunteered for scientific study and signed written consent were recruited in a PET-CT center in Huashan Hospital, Fudan University, as described before (50). The characteristics of healthy adults are listed in table S2. Primary human SVF from subcutaneous WAT used for adipocyte differentiation was obtained from underaged individuals that received plastic surgery in Ruijin Hospital, Shanghai Jiao Tong University. The human studies were approved by the Institutional Review Board of Huashan Hospital, Fudan University or Ruijin Hospital, Shanghai Jiao Tong University.
Metabolic phenotyping
Whole-body VO2, food intake, activity, and RQ were monitored by using the Promethion Continuous Indirect Calorimetry system (Sable Systems) according to the manufacturer’s protocols. Briefly, mice were single-housed in cages and acclimatized for 1 day, and then VO2 data were collected. Data were calculated by normalization to lean mass of each mouse. For induced thermogenic capacity experiments, mice were intraperitoneally injected with NE (1 mg/kg; MilliporeSigma) and were measured every 4 min. Core temperatures were measured by using rectal thermometer 508 (Physitemp) at different time points during the 3-day cold exposure.
RNA isolation, qPCR, and RNA-seq
Total RNAs were extracted from adipose tissues with the RNeasy Lipid Tissue Mini Kit (Qiagen). RNA was reverse transcribed by using the PrimeScript RT Master Mix (Takara), and qPCR was performed using the Power SYBR green PCR kit (Takara) with the ABI Prism 7500 quantitative machine (Applied Biosystems, Foster City, CA, USA). Relative expression of target gene was calculated using the comparative method by normalization to the internal control β-actin. In some experiments, data were presented as fold change by normalization to control group. Primer sequences for qPCR are listed in table S1. RNA-seq was performed on an Illumina HiSeq2000 platform. The valid data were mapped to the reference whole genome (mm10) using Bowtie. Mapped reads were counted, and fragments per kilobase of exon per megabase of library size were calculated. GO enrichment analysis was performed using goseq R Bioconductor package.
Adipose tissue digestion and flow cytometry
Murine iWAT pads were collected and digested by collagenase type II (0.5 mg/ml; MilliporeSigma) at 37°C with shaking at 100 rpm for 30 min. Digested tissues were filtered through a 100-μm nylon mesh followed by centrifugation at 1500 rpm for 5 min, and then floating adipocytes were removed for further studies, including CMKLR1 expression by flow cytometry or Il33 mRNA expression by qPCR. SVF pellets were washed once with FACS buffer [1% bovine serum albumin in phosphate-buffered saline (PBS)] and then preincubated with anti-CD16/32 and followed by staining with the appropriate fluorochrome-labeled antibodies that are listed in table S3. Cell populations in SVF including ILC2 (CD45+Lin−KLRG1+), M2 macrophages (Mφ, CD45+CD11b+F4/80+CD206+), and eosinophils (CD45+CD11b+SiglecF+) were analyzed. For detecting proliferating APs, mice were intraperitoneally injected with BrdU (100 mg/kg; Thermo Fisher Scientific) every 2 days starting from 1 day before the 3-day cold exposure, and then single-cell suspensions of SVF were stained with phycoerythrin–anti-BrdU antibody (Thermo Fisher Scientific). Samples were acquired by CyAn ADP instrument (Beckman Coulter, Miami, FL, USA) and analyzed by FlowJo software (TreeStar Inc., San Carlos, CA, USA). For some experiments, individual cell populations in SVF including leukocytes (CD45+CD31−PDGFRα−), ECs (CD31+CD45−PDGFRα−), and APs (PDGFRα+CD31−CD45− cells) were sorted by MoFlo XDP (Beckman Coulter) for measuring Il33 mRNA expression.
Preadipocyte isolation and in vitro differentiation
Single-cell suspensions of SVF were prepared from iWAT or interscapular BAT of 8-week-old male mice of different genotypes and cultured in a 100-mm dish with Advanced Dulbecco’s modified Eagle’s medium/F12 and 10% fetal bovine serum (both from Gibco) for 48 hours. For preadipocyte enrichment, the cultured SVF cells were digested by 0.25% trypsin to remove adhesive macrophages and then plated in a 100-mm dish again to grow to confluence. More than 90% of cells were PDGFRα positive as determined by flow cytometry when they were used for adipocyte differentiation as previously described (51). Briefly, enriched iWAT PDGFRα+APs were seeded in 12-well plates and treated with beige cocktail containing insulin (860 nM; Roche), dexamethasone (2 μg/ml), 3-isobutyl-1-methylxanthine (IBMX; 0.5 mM), 570 triiodothyronine (T3; 1 nM), and indomethacin (Indo; 125 mM) (all from MilliporeSigma) for 2 days followed by adding medium with insulin (850 nM) and T3 (1 nM) for another 6 days or with white cocktail containing insulin (860 nM), dexamethasone (2 μg/ml), and IBMX (0.5 mM) for 2 days followed by medium with insulin (860 nM) for another 6 days. For brown adipocyte differentiation, enriched BAT PDGFRα+APs were treated with the same cocktail used for beige adipocyte differentiation. The media was changed every other day. In some experiments, differentiated adipocytes under beige cocktail on day 8 were further stimulated with Forskolin (10 μM; Sigma-Aldrich) for 4 hours or pretreated with cAMP inhibitor Rp-8-Br-cAMPs (1 mM; Sigma-Aldrich), PKA inhibitor H89 2HCL (20 μM; Selleck), or CREB inhibitor KG-501 (5 μM; Selleck) for 1 hour, and dimethyl sulfoxide (DMSO) was used as vehicle control, followed by Forskolin stimulation. For human primary adipocyte differentiation, single-cell suspensions of SVF were prepared from human subcutaneous WAT and cultured for the enrichment of PDGFRα+APs, followed by treatment with the same adipogenic stimulation cocktail.
Western blotting and ELISA
For Western blot analysis, adipose tissues or cultured cells were homogenized in lysis buffer containing 50 mM tris-HCl, 2% SDS, protease inhibitor cocktail (Roche), and 1 mM phenylmethylsulfonyl fluoride. Total protein concentrations were measured by using a BCA protein assay kit (Thermo Fisher Scientific). Equal amounts of proteins were subjected to SDS–polyacrylamide gel electrophoresis, and the membranes were incubated with 0.1% Tween 20 (PBST) containing 5% nonfat skim milk in PBS for 1 hour at room temperature (RT). Subsequently, the membranes were incubated at 4°C overnight with primary antibodies in PBST containing 5% nonfat milk. After three washes with PBST, the membrane was incubated with a secondary antibody and developed with enhanced chemiluminescence detection system (Pierce/Thermo Fisher Scientific). For ELISA analysis, supernatants from the homogenized iWAT pad or culture of primary adipocyte differentiation were collected to determine the concentration of IL-33, IL-13, and IL-4 by ELISA kits. Chemerin concentrations were measured in sera collected from healthy adults that were recruited for PET-CT examination. The antibodies used for Western blotting and the ELISA kits are listed in table S3.
Histologic analysis
Adipose tissues or liver tissues were fixed in 4% (w/v) paraformaldehyde and embedded in paraffin to prepare tissue sections. Sections were then stained with hematoxylin and eosin (H&E) or immunohistochemically stained with UCP1 antibody (1:100; #ab10983, Abcam). Images were acquired by using an Olympus imaging system (Olympus). The density of UCP1-positive staining area was quantified by ImageJ (v1.5.2). For immunofluorescence staining, adipose tissue sections were incubated with primary antibodies against perilipin-1 (1:100; Cell Signaling Technology, #9349S) or IL-33 (1:100; R&D Systems, #AF3626) overnight at 4°C followed by staining with species-specific secondary antibodies conjugated with Alexa Fluor 488–labeled donkey anti-rabbit immunoglobulin G (IgG) antibody (1:1000; A-21206, Invitrogen) or Alexa Fluor 594–labeled donkey anti-goat IgG antibody (1:1000; A-11058, Invitrogen). All slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; 10 μg/ml) and then mounted in antifade mounting medium. Images were randomly captured with Leica DMI4000B.
Intracellular cAMP levels
Differentiated primary adipocytes were collected and lysed with 282 μl of 0.1 M HCL at RT for 20 min followed by centrifugation, and the supernatants were collected for further measurement. The levels of cAMP were quantified by a competitive enzyme immunoassay kit according to the manufacturer’s instructions (Abcam).
GTT and ITT
For the glucose tolerance test (GTT), mice were intraperitoneally injected with d-glucose (2 mg/g of body weight) after overnight fasting. For the insulin tolerance test (ITT), mice that were fasted for 4 hours during the daytime were intraperitoneally injected with human insulin (0.75 mU/g of body weight). Tail blood was drawn, and glucose levels were monitored at 0, 30, 60, 90, and 120 min after glucose injection for GTT and at 0, 15, 30, 45, and 60 min after insulin injection for ITT.
siRNA and shRNA-mediated CMKLR1 knockdown
The sequence of siRNA against human CMKLR1 gene was GCAACTTCCTTCTCATCCA. The siRNA was transfected into adipocytes using Lipofectamine RNAiMAX according to the manufacturer’s protocols (Thermo Fisher Scientific). Adenovirus-delivered shRNA targeting murine Cmklr1 was generated by cloning a designed shRNA (CCGGCATCTATGATGATGA) into the adenoviral expression vector pBlock-iT (Thermo Fisher Scientific, #K494500 and K494100) according to the manufacturer’s protocols. The vector with an insertion of a LacZ sequence (CTACACAAATCAGCGATT) was used as a negative control. Adenoviruses were amplified in 293A cells and purified using Sartorius Adenovirus Purification Kits. Adenoviruses expressing ShCmklr1 or LacZ control were subcutaneously injected to the area adjacent to inguinal fat pads of HFD-induced obese mice twice a week for 3 weeks, followed by 1 week of cold exposure.
Statistical analyses
Data are presented as means ± SEM. All analyses were performed with GraphPad Prism 7 (GraphPad). Comparisons between two groups were performed using an unpaired two-tailed Student’s t test. Multiple-group comparisons were performed using one-way analysis of variance (ANOVA) followed by a Tukey-Kramer test and two-way ANOVA followed by a Bonferroni correction to compare each group. P < 0.05 was considered statistically significant. *P < 0.05, **P < 0.01, and ***P < 0.001; ns, not significant. Raw data are listed in table S4.
Funding: This work was supported by the National Key R&D Program of China 2016YFC1305103 (to R.H.) and 2018YFA0800401 (to X.L.); Major Special Projects of the Ministry of Science and Technology 2018ZX10302207 (to Y. Lin); National Natural Science Foundation of China’s major research project 91942323 (to Y. Lin); Shanghai Science and Technology Commission 19XD1400200 (to R.H.); and National Natural Science Foundation of China grants 91642112 and 81771679 (to R.H.), 31600715 (to Y. Lin), and 31571401 and 81770861 (to X.L.). Author contributions: R.H., X.L., Y. Lin, and L.X. designed experiments. Y. Lin, L.X., Q.C., C.Z., S.L., B.L., and T.L. conducted experiments. Y. Lin, L.X., and Q.C. analyzed the data. X.W. and J.W. provided technical assistance for metabolic phenotyping experiments. D.P., W.P., and X.H. provided essential mouse lines for the study. D.P., Q.T., X.W., and J.W. discussed the results from the study. J.W., Q.Z., Y.W., and Y. Li provided human samples and analyzed the data. R.H. and X.L. supervised the study. R.H. and Y. Lin wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The RNA-seq data were deposited in the GEO database under accession number GSE133978. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.