
Abstract
The inclusion of infants in the SARS-CoV-2 vaccine roll-out is important to prevent severe complications of pediatric SARS-CoV-2 infections and to limit transmission and could possibly be implemented via the global pediatric vaccine schedule. However, age-dependent differences in immune function require careful evaluation of novel vaccines in the pediatric population. Toward this goal, we assessed the safety and immunogenicity of two SARS-CoV-2 vaccines. Two groups of 8 infant rhesus macaques (RMs) were immunized intramuscularly at weeks 0 and 4 with stabilized prefusion SARS-CoV-2 S-2P spike (S) protein encoded by mRNA encapsulated in lipid nanoparticles (mRNA-LNP) or the purified S protein mixed with 3M-052, a synthetic TLR7/8 agonist in a squalene emulsion (Protein+3M-052-SE). Neither vaccine induced adverse effects. Both vaccines elicited high magnitude IgG binding to RBD, N terminus domain, S1, and S2, ACE2 blocking activity, and high neutralizing antibody titers, all peaking at week 6. S-specific memory B cells were detected by week 4 and S-specific T cell responses were dominated by the production of IL-17, IFN-γ, or TNF-α. Antibody and cellular responses were stable through week 22. The immune responses for the mRNA-LNP vaccine were of a similar magnitude to those elicited by the Moderna mRNA-1273 vaccine in adults. The S-2P mRNA-LNP and Protein-3M-052-SE vaccines were well-tolerated and highly immunogenic in infant RMs, providing proof-of concept for a pediatric SARS-CoV-2 vaccine with the potential for durable immunity that might decrease the transmission of SARS-CoV-2 and mitigate the ongoing health and socioeconomic impacts of COVID-19.
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has infected hundreds of millions of people worldwide and caused over 3.5 million deaths since its emergence in 2019. The need for safe and effective measures to limit transmission and mitigate public health and socioeconomic impacts of SARS-CoV-2 infection has prompted unprecedented vaccine development of promising candidates. In fact two messenger RNA (mRNA) vaccines, mRNA-1273 (Moderna) (1, 2) and BNT162b2 (Pfizer-BioNTech) (3, 4), are authorized for emergency use in the United States to prevent SARS-CoV-2 infection in adults. Both are safe, and induce neutralizing antibodies and up to 95% protection from disease. Additionally, the Oxford/AstraZeneca adenovirus-based ChAdOx1 nCoV-19 (AZD1222) (5), the Johnson & Johnson Ad26.COV2.S vaccine (JNJ-78436735) (6), and the first protein-based SARS-CoV-2 vaccine, Novavax NVX-CoV2373, adjuvanted with saponin-based Matrix-M (7, 8), are approved for use in human adults.
Ethical and safety risks warrant a careful evaluation of novel vaccines in the pediatric population. Generally, vaccine testing is performed in an age de-escalation manner, starting with adults, followed by adolescents, with children and infants being last (9). The BNT162b2 mRNA vaccine is now approved for use in adolescents 12 years and older, with trials for children age 6 to 12 already underway (NCT04816643). The Moderna mRNA-1273 vaccine is close to approval for use in children 12 years and older. Importantly, Moderna and Pfizer both have initiated clinical trials that will include infant as young as 6 months (NCT04796896 and NCT04816643, respectively). The important epidemiologic impact of pediatric SARS-CoV-2 vaccination lies in limiting transmission and ease of implementation via the global pediatric vaccine schedule.
Early in the pandemic, vaccines to prevent SARS-CoV-2 infection in children were not a priority because of apparent low infection and disease rates. In most children, SARS-CoV-2 infection causes only relatively mild disease. Nonetheless, some children develop severe symptoms, such as the multisystem inflammatory syndrome, requiring hospitalization and sometimes leading to death (10–14). Importantly, severe complications of SARS-CoV-2 infection disproportionally affect children of ethnic and racial minorities, amplifying health disparities in pediatric care in the United States (15, 16). While children may transmit less efficiently than adults, virus transmission by children, even when asymptomatic, is documented (17, 18). Therefore, children have the propensity to become a major viral reservoir if pediatric vaccination stalls. The latter potential is especially concerning because new SARS-CoV-2 variants with increased transmission rates are emerging and SARS-CoV-2 has become more likely to persist on the population level. Although data are scarce, increased pediatric infection rates that appeared to coincide with the emergence of new variants, have been reported (19, 20). There is ample precedence for the beneficial impact of pediatric vaccination.
Children have suffered from the SARS-CoV-2 pandemic in many additional ways. We will not know the exact burden of the loss of education due to closure of schools and virtual learning until years from now. School closure meant that many children were deprived of social interactions, a food source, and a safe place (21–23). There has been an epidemic rise in depression and anxiety in children, and the long-term mental health burden of these diverse factors is difficult to assess. School closure also translated to loss of work for many parents, especially women. A pediatric SARS-CoV-2 vaccine could mitigate at least some of these societal costs.
These combined data emphasize the importance of getting children vaccinated for SARS-CoV-2 (24–27). However, vaccine-elicited immunity differs between adults and infants (reviewed in (28)), with impaired responses to carbohydrate antigen vaccines (29), but higher magnitude humoral immunity to subunit-based vaccines (e.g., Hepatitis B) (30) early in life. Thus, the evaluation of SARS-CoV-2 vaccine immunogenicity in infants is critical. Toward this goal, the present study was designed to evaluate the safety and immunogenicity of stabilized prefusion SARS-CoV-2 Spike vaccines delivered as lipid nanoparticle-encapsulated mRNA or an adjuvanted subunit protein in a relevant animal model. Non-human primates (NHP), including rhesus macaques (RMs), are an important model for SARS-CoV-2 studies given the similarities to humans in pathogenesis and host immune responses (31). Indeed, results from adult NHP studies of human SARS-CoV-2 vaccine candidates (32–35) strongly correlate with clinical trial outcomes, and infant NHP models of other infectious diseases closely mirror responses in human infants (36, 37). Data by our group support that infant vaccine-elicited antibody against HIV envelope can be of higher magnitude than those elicited by adults, (38) We also demonstrated that infant RMs generate high magnitude antibody responses to HIV Env protein vaccination, (39) and that peripheral and mucosal B cell responses can be boosted later in life. (40)
The primary goal of the current study was to inform human pediatric trials and aid in fast-tracking pediatric SARS-CoV-2 vaccines for inclusion in the early life vaccine schedule by providing proof-of-concept that the two SARS-CoV-2 vaccines are safe and immunogenic in infant RMs. A secondary goal of our study was to assess the durability of vaccine-induced immune responses as data are lacking on this. Despite some caveats inherent to comparisons between studies with differences in study design, the immune responses in infant RMs in response to the two SARS-CoV-2 vaccines tested appear to be within the range of vaccine-induced immune responses reported for adult RMs (32, 41) and humans (2, 42, 43).
RESULTS
Study design and vaccine safety
Infant RMs at a median age of 2.2 months, corresponding to 9-month old human infants (44), were immunized at weeks 0 and 4 with 30 μg mRNA encoding stabilized prefusion SARS-CoV-2 S-2P protein in lipid nanoparticles (mRNA-LNP; n=8) or with 15 μg S-2P mixed with 3M-052-SE, a TLR7/8 agonist in a stable emulsion (Protein+3M-052-SE; n=8) (Fig. 1, Table 1). Blood and saliva were collected before vaccination (week 0), 4 weeks after the first dose of the vaccine (week 4), 2 weeks after the vaccine boost (week 6), and at weeks 8, 14, 18 and 22. Lymph nodes were sampled at week 6 (Fig. 1).
Study Design: evaluation of immunogenicity of two SARS-CoV-2 vaccines in infant rhesus macaques. Infant rhesus macaques (median age of 2.2 months at study initiation) were immunized at 0 and 4 weeks with either 30 μg mRNA encoding S-2P (Vaccine Research Center, NIH) in lipid nanoparticles (mRNA-LNP) or 15 μg S-2P protein formulated with 3M-052 adjuvant, a TLR7/8 agonist, as a stable emulsion (3M-052-SE). Each group consisted of 8 animals. Blood and saliva samples were collected at weeks 0, 4, 6, 8, 14, 18 and 22, and lymph node biopsies were obtained at week 6.
” data-icon-position=”” data-hide-link-title=”0″>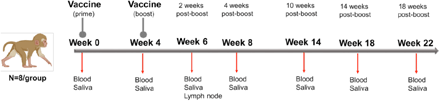
Study Design: evaluation of immunogenicity of two SARS-CoV-2 vaccines in infant rhesus macaques. Infant rhesus macaques (median age of 2.2 months at study initiation) were immunized at 0 and 4 weeks with either 30 μg mRNA encoding S-2P (Vaccine Research Center, NIH) in lipid nanoparticles (mRNA-LNP) or 15 μg S-2P protein formulated with 3M-052 adjuvant, a TLR7/8 agonist, as a stable emulsion (3M-052-SE). Each group consisted of 8 animals. Blood and saliva samples were collected at weeks 0, 4, 6, 8, 14, 18 and 22, and lymph node biopsies were obtained at week 6.
Animals were monitored daily for adverse events. No local injection site or systemic adverse reactions were observed. Animals had normal blood metrics (Supplementary Table S1). Importantly, the animals gained weight consistent with adequate fluid and nutrition uptake and with normal infant growth (Supplementary Fig. S1) (45). A major safety concern in the implementation of a pediatric SARS-CoV-2 vaccine stems from adverse events observed with an earlier Respiratory Syncytial Virus vaccine, that were partially attributed to T helper 2 (TH2)-biased responses (46, 47). We therefore tested plasma samples prior to or following immunization for changes in TH2 (IL-4, IL-13) or TH1 (IL-2, IFN-γ) cytokines (Fig. S2). Animal RM3 of the protein group (Table 1) tested positive for all cytokines at both time points; cytokines were also detected in the mRNA-LNP recipient RM11 at week 0, but fell below the limit of quantification by week 6. In general, neither vaccine appeared to induce systemic TH2 or TH1 responses (Fig. S2), further supporting a good safety profile.
Infant plasma and salivary antibody responses to SARS-CoV-2 vaccination
Plasma IgG binding to the S-2P protein was observed after the vaccine prime for both mRNA-LNP and Protein+3M-052-SE vaccines and increased after the second immunization (Fig. 2). Although antibody levels dropped by week 8, there was only a slight decline of S-2P-specific IgG from week 8 to week 22 for mRNA-LNP (median AUC and 95% confidence intervals: 7.0 [6.2, 8.3] to 5.4 [4.2, 7.7]) or Protein+3M-052-SE (11.0 [9.5, 11.7] to 9.4 [8.6, 10.5] vaccinees (Fig. 2A). Longitudinal S-specific plasma IgG data reported as EC50 or endpoint dilution titers followed a similar trend (Fig. S3). As the D614G virus variant had become dominant in the USA at the study initiation, we confirmed that IgG binding to S-2P D614G was similar to that of D614 at weeks 6 and 14 (Fig. S4). Plasma IgG antibodies were directed against multiple spike domains and persisted throughout the study, with robust binding to Spike regions 1 and 2 (S1, S2), Receptor binding domain (RBD), and N-terminal domain (NTD) (Fig. 2B). RBD-specific salivary IgG from mRNA-LNP recipients peaked at a median of 16.6 ng RBD-specific IgG per μg of total IgG at week 6 (Fig. 2C). In the Protein+3M-052-SE group, median salivary RBD-specific IgG peaked at 98.2 ng/μg IgG after the second vaccination and remained detectable throughout the study (Fig. 2C). Saliva RBD-specific IgA responses were much lower (Fig. S5). Spike-specific IgM and IgA in plasma were low or undetectable (Fig. S5). In the mRNA-LNP group, IgM increased through week 14, but the absorbance readings for IgM were considered low, measured at a 1:10 sample dilution (Fig. S5).
Plasma and saliva were collected before vaccination (week 0), at week 4 – prior to the 2nd dose-, two weeks post 2nd dose (week 6), and at weeks 8, 14, 18 and 22 from infant RM vaccinated with 30 μg mRNA encoding S-2P spike protein in lipid nanoparticles (n=8; red) or with 15 μg prefusion SARS-CoV-2 S-2P spike protein formulated with 3M-052 adjuvant (n=8; blue). (A): S-2P protein-specific antibody responses were measured by enzyme-linked immunosorbent assay (ELISA). Serial dilutions of plasma starting at 1:40 were assayed for IgG binding to SARS-CoV-2 spike. Data are reported as log10 area under the curve (AUC) values. (B): Antibody epitope specificity measured by BAMA. Plasma was diluted 1:10,000 to measure binding to different domains of the spike protein, including the full-length S protein, S1, RBD, NTD, and S2. Binding antibody responses are reported as log10 transformed mean fluorescence intensity (MFI) after subtraction of background values. (C): Salivary RBD-specific IgG was measured by binding antigen multiplex assay (BAMA) using serial dilutions of saliva and responses are reported as RBD-specific IgG (ng) / total IgG (μg). Different symbols represent individual animals (Table 1). Arrows in Panel A indicate times of immunizations.

Plasma and saliva were collected before vaccination (week 0), at week 4 – prior to the 2nd dose-, two weeks post 2nd dose (week 6), and at weeks 8, 14, 18 and 22 from infant RM vaccinated with 30 μg mRNA encoding S-2P spike protein in lipid nanoparticles (n=8; red) or with 15 μg prefusion SARS-CoV-2 S-2P spike protein formulated with 3M-052 adjuvant (n=8; blue). (A): S-2P protein-specific antibody responses were measured by enzyme-linked immunosorbent assay (ELISA). Serial dilutions of plasma starting at 1:40 were assayed for IgG binding to SARS-CoV-2 spike. Data are reported as log10 area under the curve (AUC) values. (B): Antibody epitope specificity measured by BAMA. Plasma was diluted 1:10,000 to measure binding to different domains of the spike protein, including the full-length S protein, S1, RBD, NTD, and S2. Binding antibody responses are reported as log10 transformed mean fluorescence intensity (MFI) after subtraction of background values. (C): Salivary RBD-specific IgG was measured by binding antigen multiplex assay (BAMA) using serial dilutions of saliva and responses are reported as RBD-specific IgG (ng) / total IgG (μg). Different symbols represent individual animals (Table 1). Arrows in Panel A indicate times of immunizations.
Function of vaccine-induced plasma antibodies
We next evaluated the capacity of plasma antibodies to block entry of SARS-CoV-2 into human cells using a RBD-ACE2 blocking assay at 1:10 and 1:40 plasma dilutions. Antibodies elicited by the mRNA-LNP vaccine completely blocked RBD-ACE2 interaction at week 6 at 1:10 and 1:40 dilutions. Greater than 80% blocking was achieved at 1:10 dilution until week 14, dropping for some animals after week 18 (Fig. 3A). The Protein+3M-052-SE vaccine induced antibodies that mediated 100% RBD-ACE2 blocking after the second immunization (Fig. 3A). At a 1:40 plasma dilution, the RBD-ACE2 blocking function decreased over time, but was still detectable at week 22 (Fig. S6).
SARS-CoV-2 vaccine-elicited functional antibody responses in infant rhesus macaques. (A): The capacity of plasma antibodies to mediate blocking of the RBD-ACE2 interaction was measured with an ELISA-based ACE2 blocking assay at 1:10 plasma dilution. Data are reported as %ACE2 blocking. (B-C): Neutralization capacity was measured using Spike D614G-pseudotyped viruses in 293T/ACE2 cells (B) and whole virus (D614G) assay with Vero E6 cells (C); results are expressed as reciprocal 80% inhibitory dilution (ID80). Grey dotted lines represent detection cut-off. Different symbols represent individual animals (Table 1). Longitudinal data for each animal in the mRNA-LNP (red) or Protein+3M-052-SE group (blue) are represented by separate lines. Arrows in Panel C indicate times of immunizations.

SARS-CoV-2 vaccine-elicited functional antibody responses in infant rhesus macaques. (A): The capacity of plasma antibodies to mediate blocking of the RBD-ACE2 interaction was measured with an ELISA-based ACE2 blocking assay at 1:10 plasma dilution. Data are reported as %ACE2 blocking. (B-C): Neutralization capacity was measured using Spike D614G-pseudotyped viruses in 293T/ACE2 cells (B) and whole virus (D614G) assay with Vero E6 cells (C); results are expressed as reciprocal 80% inhibitory dilution (ID80). Grey dotted lines represent detection cut-off. Different symbols represent individual animals (Table 1). Longitudinal data for each animal in the mRNA-LNP (red) or Protein+3M-052-SE group (blue) are represented by separate lines. Arrows in Panel C indicate times of immunizations.
Neutralizing antibodies, measured by a pseudovirus assay, were detected in all mRNA-LNP vaccinated animals and in 7 of 8 animals of the Protein+3M-052-SE group after the first vaccination, and further increased after the boost. Peak median infectious dose 80 (ID80) titers were observed at week 6 and reached 1,179 in mRNA-LNP and 13,687 in Protein+3M-052-SE animals (Fig. 3B). Similar to S-specific plasma IgG levels, and consistent with antibody kinetics after vaccination, median ID80 neutralization titers decreased following the peak response. Neutralization titers appeared to stabilize around week 18, and median ID80 titers at week 22 remained 2.6-fold or 14.2-fold higher in the mRNA-LNP or Protein+3M-052-SE group, respectively, than after the first vaccination (week 4) (Fig. 3B). Although definite immune correlates of protection after vaccination still need to be conclusively determined, we modeled a bi-phasic antibody decline for both vaccines to reach a putative protective neutralizing Ab titer, estimated at a reciprocal dilution of 100 (41, 48). Applying this assumption, mRNA-LNP vaccinees maintained a protective neutralizing Ab titer for approximately 46 weeks and protein vaccine recipients for approximately 74 weeks (Fig. S7).
Neutralizing antibody kinetics in the whole virus neutralization assay followed a similar trend (Fig. 3C). The animal (RM 1) of the Protein+3M-052-SE group that did not have detectable neutralizing antibodies by week 4 in the pseudovirus neutralization assay, also only had a slight increase in the ID80 neutralizing antibody titer using the live virus assay (week 0: 129; week 4: 223). Overall, ID50 and ID80 titers of both assays strongly correlated with each other (r = 0.644, p=0.008 or r=0.785, p=0.0005, respectively Fig. S8). However, infant RMs already had low titer neutralizing antibodies at day 0 when we applied the whole virion neutralization assay (Fig. 3C). As infants could have passively acquired antibodies from their mothers, we measured neutralizing antibodies in the sera of their unvaccinated dams. Although detectable, neutralizing antibody titers in dams did not correlate to infant neutralization titers before (ID80: r=0.011, p=0.96) or after vaccination (ID80: r=-0.056, p=0.83) (Fig. S9), most likely because infant rhesus macaques were already 2 months old at study initiation and maternal antibodies had waned.
Vaccine-elicited B cell responses
Consistent with the induction of plasma S-specific IgG, S-specific memory CD27+ B cell were detectable in the blood of both vaccine groups at week 4 and peaked at week 6 (median: 0.75% mRNA-LNP; 3.12%, Protein+3M-052-SE). At week 14, median memory B cell frequencies were lower (0.19% and 0.13% in mRNA-LNP or Protein+3M-052-SE vaccinees, respectively), but did not differ from those at week 4 (Fig. 4A). In lymph nodes (LNs), we measured total germinal center (GC) B cells and memory B cells and then determined the number of S-specific memory B cells (Fig. 4B). In both vaccine groups robust memory B cell populations (median 3.78% mRNA-LNP and 1.96% Protein-3M-052) were present (Fig. 4B). Induction of S-specific B cells was confirmed by assessing S-specific antibody secreting cells (ASC). In the blood, peak responses were observed at week 6 in the mRNA-LNP group (median: 154 ASC/million cells) and at week 8 in the Protein+3M-052-SE group (median: 85 ASC/106 cells) (Fig. 4C). In draining LNs, mRNA-LNP and Protein+3M-052-SE vaccinees had median 6 or 952 ASC/106 cells, respectively (Fig. 4D).
(A) CD20+CD27+ memory B cells that co-stained with fluorochrome-conjugated SARS-CoV-2 spike protein in mRNA-LNP (red) or Protein+3M-052-SE (blue) vaccinees in blood. Frequencies are expressed as percent of total memory B cells. The gating strategy is provided in Supplementary Figure S14. (B): In lymph nodes, we determined total Bcl-6+Ki-67+ Germinal Center (GC) B cells and CD27+ memory B cells as percent of total CD20+ B cells (left panel) (see Fig. S15 for gating strategy) and also the percent of S-specific memory B cells (right panel). (C) Antibody secreting cell (ASC) as measured by B cell ELISpot in PBMC from mRNA-LNP or Protein+3M-052-SE vaccinees, while (D) is showing mRNA-LNP and Protein+3M-052-SE ASC responses, respectively, in LN at week 6. Different symbols represent individual animals (Table 1). Solid lines represent median values.
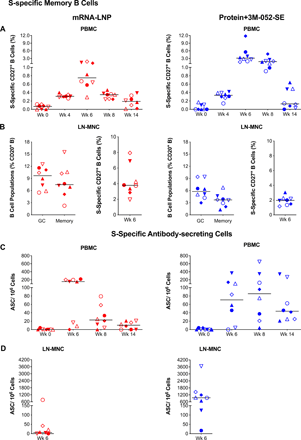
(A) CD20+CD27+ memory B cells that co-stained with fluorochrome-conjugated SARS-CoV-2 spike protein in mRNA-LNP (red) or Protein+3M-052-SE (blue) vaccinees in blood. Frequencies are expressed as percent of total memory B cells. The gating strategy is provided in Supplementary Figure S14. (B): In lymph nodes, we determined total Bcl-6+Ki-67+ Germinal Center (GC) B cells and CD27+ memory B cells as percent of total CD20+ B cells (left panel) (see Fig. S15 for gating strategy) and also the percent of S-specific memory B cells (right panel). (C) Antibody secreting cell (ASC) as measured by B cell ELISpot in PBMC from mRNA-LNP or Protein+3M-052-SE vaccinees, while (D) is showing mRNA-LNP and Protein+3M-052-SE ASC responses, respectively, in LN at week 6. Different symbols represent individual animals (Table 1). Solid lines represent median values.
In addition, we analyzed LN samples for canonical T follicular helper (TFH) (CD185/CXCR5+CD279/PD-1+) cells and for IL-4 or IL-21 producing CD4+ T cells that support GC B cell differentiation (Fig. 5A). SEB-activated TFH (CD185+CD279+CD134+CD137+) (49) (Fig. 5B) were categorized into TFH1, TFH2 and TFH17 subsets based on expression of CXCR3/CD183 and/or CCR6/CD196 (50) (Fig. 5C). We found no correlations between bcl6+Ki67+ GC B cells or bcl6+TFH (Fig. S10A, B), potentially because we assessed these cell populations in an antigen non-specific manner. However, CD4+IL21+ cells correlated with pseudovirus neutralizing antibody ID50 at week 6 (r=0.749, p=0.001) (Fig. S10C).
Immunophenotype of lymph node T cell population two weeks post-boost. (A): CD4+ T-cells positive for IL-4 or IL-21, follicular T helper cell (Tfh) markers CD279/PD-1 and CD185/CXCR5, or Bcl6+ Tfh were measured and are represented as percent of total LN CD4+ T cells. The gating strategy for these panels is provided in Figure S15. (B): SEB activated Tfh frequencies assessed by the Activation-Induced Marker (AIM) Assay: CXCR5+CD185+ cells that co-expressed CD134 and CD137 for Protein+3M-052-SE and mRNA-LNP, respectively. (C) The frequency of CXCR5+CD185+CD134+CD137+CXCR3+CD196– Tfh1, CXCR3–CCR6– Tfh2 and CXCR3–CD196+ Tfh17 cells is shown. The gating strategy for these populations is given in Fig. S17. Red symbols: mRNA-LNP, blue symbols: Protein+3M-052-SE. Different symbols represent individual animals (Table 1). Solid lines define the median.
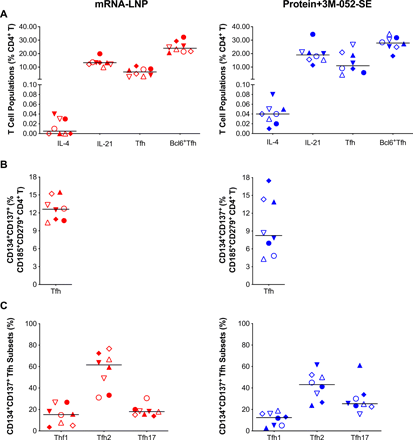
Immunophenotype of lymph node T cell population two weeks post-boost. (A): CD4+ T-cells positive for IL-4 or IL-21, follicular T helper cell (Tfh) markers CD279/PD-1 and CD185/CXCR5, or Bcl6+ Tfh were measured and are represented as percent of total LN CD4+ T cells. The gating strategy for these panels is provided in Figure S15. (B): SEB activated Tfh frequencies assessed by the Activation-Induced Marker (AIM) Assay: CXCR5+CD185+ cells that co-expressed CD134 and CD137 for Protein+3M-052-SE and mRNA-LNP, respectively. (C) The frequency of CXCR5+CD185+CD134+CD137+CXCR3+CD196– Tfh1, CXCR3–CCR6– Tfh2 and CXCR3–CD196+ Tfh17 cells is shown. The gating strategy for these populations is given in Fig. S17. Red symbols: mRNA-LNP, blue symbols: Protein+3M-052-SE. Different symbols represent individual animals (Table 1). Solid lines define the median.
Spike protein-specific T cell responses
In some animals, S-specific CD4+ T cell responses in PBMC were detected as early as week 4, with all animals producing at least a single cytokine by week 6 (Fig. 6; see Fig. S16 for gating strategies). At week 14, CD4+ T cells of mRNA-LNP vaccinated animals produced IL-2, IFN-γ, IL-17, and TNF-α responses (Fig. 6D), whereas IL-17 and IFN-γ CD4+ T cell responses dominated in Protein+3M-052-SE vaccinees (Fig. 6D). Multifunctional CD4+ T cells co-produced IL-17 and IFN-γ (Fig. S11), suggesting a TH1/TH17 biased response. Although we did not measure IL-4 production, and limited cell numbers – typical for pediatric samples – prevented us from repeating the assay, it should be reiterated that neither of the vaccines caused a rise in systemic TH2 cytokines (Fig. S2). S-specific peripheral blood CD8+ T cell responses appeared less robust than CD4+ T cell responses and only single-cytokine positive were elicited, but all animals produced at least a single cytokine in response to antigen stimulation (Fig. S12). In LNs, 6 of 8 and 8 of 8 animals in the mRNA-LNP or Protein+3M-052-SE group had S-specific CD4+ T cell responses, respectively. In addition, 8 of 8 mRNA-LNP vaccinees and 7 of 8 Protein+3M-052-SE vaccinees mounted S-specific CD8+ T cell responses in LN at week 6 (Fig. S13).
Intracellular cytokine staining for IL-2, IL-17, IFN-γ, and TNF-α (indicated at the x-axis in Panel D) was performed on PBMC at weeks 4 (A), 6 (B), 8 (C), and 14 (D) to assess T-cell responses to a peptide pool encompassing the entire SARS-CoV-2 spike protein (see Fig. S16 for gating strategy). Responses detected in mRNA-LNP recipients are displayed in red and cytokine responses from Protein+3M-052+SE vaccinees are in blue. The dashed lines represent week 0 values plus 2 standard deviations and define the cutoff for positive cytokine responses. Different symbols represent individual animals (Table 1).
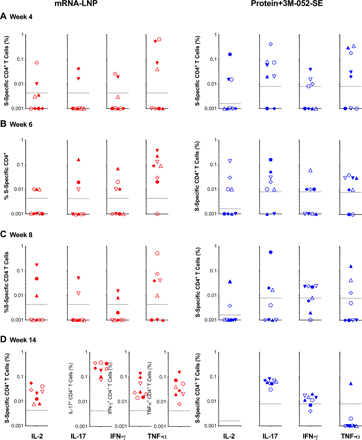
Intracellular cytokine staining for IL-2, IL-17, IFN-γ, and TNF-α (indicated at the x-axis in Panel D) was performed on PBMC at weeks 4 (A), 6 (B), 8 (C), and 14 (D) to assess T-cell responses to a peptide pool encompassing the entire SARS-CoV-2 spike protein (see Fig. S16 for gating strategy). Responses detected in mRNA-LNP recipients are displayed in red and cytokine responses from Protein+3M-052+SE vaccinees are in blue. The dashed lines represent week 0 values plus 2 standard deviations and define the cutoff for positive cytokine responses. Different symbols represent individual animals (Table 1).
Overall, these data suggested that infant RMs can mount robust antibody, including high titer neutralizing and ACE2 blocking antibodies, and T cell responses to SARS-CoV-2 vaccines. Vaccine-induced immune responses persisted for 22 weeks or 18 weeks after the first and second vaccine dose, respectively, results consistent with vaccine-induced memory.
DISCUSSION
Several SARS-CoV-2 vaccines have demonstrated safety, immunogenicity and protection in animal studies (8, 32, 51, 52) and in clinical trials with human adults (1, 5, 6), and subsequently received authorization for emergency use in human adults. Considering the enormous health and socioeconomic impact of the SARS-CoV-2 pandemic on all age groups, clinical trials to test the safety of SARS-CoV-2 vaccines in adolescents and children have been initiated. In fact, the Pfizer-BioNTech mRNA vaccine is already approved for adolescents 12 years and older, Moderna’s vaccine (NCT04796896) has recently been reported to be 96% effective in adolescents ages 12-17, and Novavax (NCT04611802) has an ongoing trial to test its vaccine this age group. However, data regarding the safety and immunogenicity of SARS-CoV-2 vaccines in young infants are still lacking.
We reasoned that validating safety and immunogenicity of SARS-CoV-2 vaccines in infant rhesus macaques would provide beneficial information supporting the initiation of pediatric vaccine trials down to 6 months of age. Here, we present preclinical data demonstrating that infant rhesus macaques develop strong, durable humoral and cellular responses in the absence of adverse events following vaccination with a preclinical version of the Moderna mRNA-1273 vaccine or with stabilized prefusion S-2P SARS-CoV-2 protein mixed with 3M-052-SE, a TLR7/8 agonist in squalene emulsion. We selected the mRNA-LNP vaccine expressing S-2P because at the initiation of the study mRNA vaccines were quickly progressing through phase 3 clinical studies toward approval for human use, with EUA approval in the US in late 2020. We therefore considered this to be the class of vaccines most likely to be among the first vaccines to eventually progress for testing in the pediatric population. The 3M-052-SE adjuvant was chosen for the S-2P protein vaccine because this adjuvant has proven effective in eliciting high magnitude antibody responses to other vaccines in infant rhesus macaques (39, 53).
We assessed infant vaccine-induced immune responses over 22 weeks, analogous to adult clinical trials (1, 2). Both vaccines elicited plasma antibodies dominated by IgG and recognized all Spike protein domains. Binding antibodies persisted throughout the study in all animals of both groups. Interestingly, RBD-specific IgG was also detected in saliva, especially in animals of the protein vaccine group, and remained detectable throughout the study, similar to what has been observed after human natural infection (54). Moreover, 7 of 8 Protein+3M-052-SE vaccinated animals had RBD-specific IgA after the 2nd immunization. The overall low induction of S-2P- and RBD-specific IgA by vaccination might be attributed to the delayed development of mucosal IgA compared to IgG responses in human and rhesus infants (55–57). Vaccine-induced neutralizing antibody kinetics paralleled those observed for plasma binding antibodies and persisted for the duration of the study. At week 22, 18 weeks after the 2nd dose, median ID50 titers in the pseudovirus assay still exceeded 103 for the protein vaccine group and 102 for the mRNA group.
The immune correlates of protection against SARS-CoV-2 infection and disease still need to be conclusively determined (58, 59). To address this question, McMahan et al. (48) adoptively transferred plasma IgG from adult SARS-CoV-2 convalescent rhesus macaques and determined that a S protein-specific reciprocal endpoint dilution ELISA titer of 400 and pseudovirus neutralizing antibody (ID50) titers of approximately 50 can protect adult rhesus macaques against combined intratracheal and intranasal SARS-CoV-2 challenge (48). Applying these criteria to our results, we hypothesized that pediatric vaccination could provide protection against SARS-CoV-2 infection. However, we purposely decided to delay the challenge of our animals until one year post vaccination to better determine the long-term persistence of vaccine-induced neutralizing antibodies, which is especially important to a pediatric vaccine for a disease that affects all age groups. These data will be reported in a follow up study. Furthermore, the drop in ACE2-blocking function in some animals of the mRNA-LNP group raises the possibility that some of our vaccinated animals might develop disease, whereas others would be protected. Such an outcome would allow us to define immune correlates of protection, including the protective titer of neutralizing antibodies against infection and from severe disease, but also enable us to assess the contribution of other vaccine-induced immune responses (e.g., T cell responses) to protection.
Early life immunity is associated with TH2-biased T cell responses (60), and TH2 responses have been linked to vaccine-associated enhanced respiratory disease in the context of protein or inactivated virus vaccines (6, 61, 62). We observed a TH1/ TH17-skewed cytokine profile in circulating S-specific T cells. Our finding of peak S-specific T cell responses 10 weeks post-boost are not unexpected. T cell reactivity in convalescent COVID-19 patients have augmented frequency and potency 100 days post-recovery, even as S-specific antibody waned (63). Indeed, ~93% of “exposed asymptomatic” patients possess SARS-CoV-2-specific memory T cell subsets in the absence of seroconversion (64). Higher TFH2 frequencies compared to TFH1 do not necessarily imply a TFH2-bias as TFH17 cells were also detected, and TFH were not assessed for antigen specificity. We found no evidence of systemic TH2 cytokines prior to or following immunization in either vaccine group, corroborating previous findings in adult macaques (32) and adult humans (42) that also demonstrated low level or absent TH2-mediated responses. IL-4 and IL-21 producing TFH are required for the induction of GC B cells in LNs (65), indispensable for B cell and antibody affinity maturation, and GC reactions are important for positive outcomes in COVID-19 patients (65). Indeed, the persistence of antibody was paralleled by sustained S-specific B cell responses. The induction and persistence of S-specific B cell clones described here may be key to protection against re-infection (66). In the current study, S-specific CD8+ T cell responses were lower compared to CD4+ T cell responses in the same animals. As CD8+ T cell response play an important role in the control of virus replication, future studies need to determine the contribution of vaccine-induced cytotoxic T cell responses to protection against SARS-CoV-2 infection.
Overall, the adjuvanted protein vaccine seemed to elicit stronger immune responses compared to the mRNA vaccine. We can only speculate why the vaccine-induced immune responses were different between the Protein+3M-052-SE and the mRNA-LNP regimens. First, we do not know the exact amount of protein expressed by the mRNA-LNP vaccine in vivo. Second, we and others have previously demonstrated that the TLR7-8-based adjuvant 3M-052 is highly effective in enhancing antibody responses in the rhesus macaque model (39, 53). TLR7/8 agonists, in contrast to most other TLR agonists, promote potent IL-12 responses by antigen-presenting cells (67–69) that in neonates and young infants are normally biased toward IL-23 production (70). Therefore, the priming of T helper cells and antibody responses is assumed to be improved by the inclusion of the 3M-052-SE adjuvant. Nonetheless, the data do not imply that the mRNA-LNP vaccine was less successful in inducing immune responses compared to responses observed in human clinical trials or in adult NHP. In fact, S-specific T and B cell responses to the mRNA-LNP vaccine persisted throughout the study period.
It is difficult to directly compare the results of the current study with published data from human clinical trials or from adult NHP studies. We are not aware of published data using the Protein+3M-052-SE vaccine for SARS-CoV-2 in NHP. Results from human and NHP studies with the mRNA-1273 vaccine are listed in Table 2 (2, 32, 41–43). It should be noted that the various studies differed in their design. Keeping this caveat in mind, the magnitude, quality, and durability of the mRNA-LNP vaccine-induced responses in infant macaques appear to be within the range of those observed in adult NHP and human studies.
We demonstrated that infant rhesus macaques mount strong and durable responses to mRNA-LNP and protein-based SARS-CoV-2 vaccines that were comparable to adults without adverse reactions. These promising results endorse clinical translation of SARS-CoV-2 vaccines to early life populations. Even when the majority of adults and adolescents have been vaccinated and herd immunity is achieved, globally about 140 million infants are born each year (71) and they will be naïve and susceptible to SARS-CoV-2 once passively transferred maternal antibodies wane. Being cautiously optimistic, a multivalent SARS-CoV-2 vaccine effective against major circulating viral variants appears feasible and could become part of the standard pediatric vaccine program.
MATERIALS and METHODS
Study Design
The objective of this study was to provide proof-of-concept that young infants (<1 year) could mount functional and durable neutralizing antibody responses and T cell responses to SARS-CoV-2 vaccination. Considering the extent of the SARS-CoV-2 pandemic, the rapid emergence of new viral variants with increased transmission rates with yet unknown impact on pediatric infections, our goal was to inform human SARS-CoV-2 vaccine age de-escalation studies to fast-track vaccine implementation in the pediatric population. Utilizing the highly relevant infant rhesus macaque model, we tested the safety and immunogenicity of a preclinical version of the mRNA 1273 SARS-CoV-2 vaccine (n=8) and a stabilized prefusion SARS-CoV-2 S-2P spike (S) protein vaccine mixed with the 3M-052 adjuvant in stable emulsion (n=8) (Fig. 1; Table 1). S-specific cellular responses and antibody responses were monitored for 22 weeks.
Animals
Infant male (n=8) and female (n=8) rhesus macaques (Macaca mulatta; RM) of Indian-origin from the California National Primate Research Center (CNPRC, Davis, CA) breeding colony (negative for type D retrovirus, simian immunodeficiency virus, simian lymphocyte tropic virus type 1 and SARS-CoV-2), were enrolled at a median age of 2.2 months and randomly assigned into two groups (Table 1). Infants were housed with their dams until about 6 months of age, and then weaned and pair-housed. Animal care was in compliance with the “Guide for Care and Use of Laboratory Animals” by the Institute for Laboratory Animal Research. Animal procedures were approved by the UC Davis Institutional Animal Care and Use Committee prior to study initiation. All procedures were performed under anesthesia (ketamine, 10 mg/kg body weight, intramuscularly [IM]). Blood, saliva and lymph nodes (LN) were collected and processed as described (72).
Preparation of the SARS CoV2 Spike protein
The CoV2 Spike (73) (2019-nCoV) protein was transiently expressed in Freestyle HEK293 cells (Thermo Scientific) by first diluting the plasmid in Opti-MEM-1 (Gibco) medium; 0.8mg of plasmid in 25mL of Opti-MEM-1 for each liter of cells transfected and sterile filtered using a 0.22 μm filter. 1 mL of 293fectin (Gibco) was diluted in 25 mL of Opti-MEM-1 and allowed to incubate at room temperature for 5 ± 1 min. The plasmid and the 293fectin mixtures were combined and swirled to mix, then incubated at room temperature for 25 ± 5 min. During the incubation, the Freestyle HEK293 cells were diluted to 1.25×106 cells/ mL in a 2-liter Corning flask. At the end of the 25 ± 5 min incubation of the 293fectin and plasmid mixtures, 50mL of the transfection mixture was added to each flask of diluted cells with gentle agitation of the flask during addition. The cell + transfection mixture was then split between two 2-liter Corning flasks for a final volume of approximately 500mL each and placed on a platform shaker in a humidified incubator at 37°C with 8% CO2. The shaker speed was set to 120rpm and the flasks allowed to incubate for 6 days. At the end of the 6-day incubation, the flasks containing the transfected cells were removed from the incubator and the contents of the flasks were aseptically transferred to 500mL Corning centrifuge tubes. The supernatant was clarified by centrifugation and subsequently filtered using a 0.8μm filter bottle system. Keeping the clarified and filtered supernatant on ice, the supernatant was concentrated using a Sartorius Vivaflow 200 30kDa TFF system. In preparation for the purification of the CoV2 Spike protein, 4mL of Strep-Tactin Resin (iba Life Science) was placed in a conical tube. The concentrated supernatant was transferred to the conical tube containing the Strep-Tactin resin and allowed to bind with gentle agitation. The supernatant and resin were then transferred to a polypropylene column for gravity-flow chromatography (BioRad). Once the resin had settled in the column the resin was washed and the protein eluted following instructions supplied by the vendor (iba Life Science). The eluate was buffer-exchanged into 2mM Tris and 200mM Sodium Chloride storage buffer using Millipore Centrifugal filters. The resulting material was finally filtered through a 0.2um syringe filter (Pall) and stored in storage buffer. Additional purification by size exclusion chromatography over a Superose 6 column was performed to increase purity.
Vaccines
The SARS-CoV-2 stabilized prefusion Spike (S-2P) mRNA-LNP vaccine was provided by Moderna, Inc. and the Vaccine Research Center (NIH) provided the S-2P protein. The vaccine regimen and specimen collection are outlined in Fig. 1. In humans, the dose of the Moderna mRNA-1273 vaccine is 100 μg. However, even a 25 μg dose has proven immunogenic and was associated with fewer side effects compared to the 100 μg in older adults (42). In adult RMs, a 10 μg dose regimen induced significantly lower neutralizing antibody responses than the 100 μg vaccine (32). Thus, balancing immunogenicity and safety, we decided to immunize infant RMs in the mRNA-LNP vaccine group IM at weeks 0 (quadriceps) and 4 (biceps) with 30 μg mRNA encoding S-P2 protein in lipid nanoparticles (mRNA-LNP), administered in 0.1 mL phosphate-buffered saline. Note that the vaccine was stored at -80°C until just prior to the immunization. Infant RMs in the protein vaccine group were injected IM with15 μg S-2P protein mixed with 3M-052-SE, an adjuvant formulation consisting of 10 μg of the synthetic TLR7/8 agonist 3M-052 in a 2% v/v squalene-in-water emulsion (Protein+3M-052-SE) in 0.5 mL divided across the left and right quadriceps (week 0) or biceps (week 4).
Plasma IgG ELISA
IgG binding to the stabilized SARS-CoV-2 spike protein S-2P with or without the D614G mutation was measured in plasma using enzyme-linked immunosorbent assay (ELISA) as previously described (74). 384-well plates were coated overnight with 2 ug/mL of spike protein produced by the Protein Production Facility (PPF) at the Duke Human Vaccine Institute. Plates were then blocked with assay diluent (phosphate-buffered saline containing 4% whey, 15% normal goat serum, and 0.5% Tween 20). 10 serial 4-fold dilutions of plasma starting at 1:40 were added to the plates and incubated for 1 hour, followed by detection with a horseradish peroxidase (HRP)-conjugated antibody mouse anti-monkey IgG (Southern Biotech). The plates were developed by using an ABTS-2 [2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid)] peroxidase substrate system (KPL) and absorbance was read at 450 nM with a Spectramax Microplate Reader (Molecular Devices). Results were calculated as area under the curve (AUC) and EC50 values. AUC values were calculated using the Trapezoidal rule. EC50 values were calculated by fitting a 4-parameter logistic function using nonlinear regression. Pooled Non-Human Primate Convalescent Serum to SARS-CoV-2 (BEI Resources NR-52401) was used in all assays to ensure inter-assay reproducibility, but standard curves were not developed given the lack of a rhesus macaque-specific IgG reagent of known concentration. IgM and IgA binding to S-2P was measured in a single 1:10 diluted plasma sample following the same ELISA protocol but using as detection antibody anti-Human IgM HRP (Jackson Immunoresearch) for IgM detection and 10F12-biotin (NHP Reagent Resource) with Streptavidin-HRP (Pierce) for IgA detection. Results are expressed as OD450.
Measurement of Salivary S-specific IgG and IgA
Saliva was collected with absorbent Merocel sponges (Beaver Visitec) by placing a sponge between the cheek and gum in the back of the mouth for 5 min. Secretions were eluted by centrifugation at 18,000 g and 4°C after addition of 50μl of PBS containing protease inhibitors (75), 1% TritonX-100, 1% BSA, 0.05% azide, and 0.05% Tween-20 to sponges. A customized binding antigen multiplex assay (BAMA) was used to measure IgG or IgA antibodies to SARS CoV-2 recombinant receptor binding domain protein (RBD; generously provided by Dr. Wrammert, Emory University, Atlanta, GA) and S2 extracellular domain (SinoBiologicals #40590-V08B, Wayne, PA). Briefly, proteins were dialyzed in PBS and conjugated to Bioplex Pro carboxylated magnetic beads (Bio-Rad, Hercules, CA) using N-hydroxysulfosuccinimide and ethylcarbodiimide as described (76). Serial dilutions of standard and centrifuged salivary secretions in PBS containing 1% TritonX-100, 1% BSA, 0.05% azide, and 0.05% Tween-20 were mixed with RBD+S2 beads overnight at 1100rpm and 4°C using a plate mixer. The IgG standard was a cocktail of anti-S1 RBD (Genscript #HC2001) and anti-S2 (SinoBiologicals #40590-D001) humanized IgG monoclonal antibodies. The standard for IgA assays was a pooled serum from infected rhesus macaques (77) that had been calibrated relative to the previously mentioned monoclonal antibodies. The following day, beads were alternately washed using a Bio-Rad BioPlex wash station and treated for 30min with 2μg/ml biotinylated affinity-purified goat antibody to human γ chain (SouthernBiotech Associates, Birmingham, AL) or clone IgA5-3B mouse anti-monkey IgA (Bio-Rad) followed by 1/400 avidin-phycoerythrin (Southern Biotechnology Associates). A Bio-Rad Bioplex 200 and BioManager software were used to measure fluorescent intensity and construct standard curves for interpolation of antibody concentrations in test samples. Concentrations of antibody measured in a Luminex-based binding antibody multiplex assay (BAMA) were normalized relative to the total IgG and IgA measured by ELISA as described (75) using plates coated with goat anti-monkey IgG or IgA and the secondary antibodies above.
Plasma S-specific IgG Epitope Mapping
SARS-CoV-2 antigens, including whole spike (produced by PPF), S1 (Sinobiological cat# 40591-V08H), S2 (Sinobiological cat# 40590-V08B), RBD (Sinobiological cat# 40592-V08H) and NTD (Sinobiological cat# 40591-V49H) were conjugated to Magplex beads (Bio-Rad, Hercules, CA). The conjugated beads were incubated on filter plates (Millipore, Stafford, VA) for 30 min before plasma samples were added. Plasma samples were diluted in assay diluent (1% dry milk, 5% goat serum, and 0.05% Tween 20 in 1× phosphate buffered saline, pH 7.4.) at a 1:10,000-point dilution. Beads and diluted samples were incubated for 30 min with gentle rotation, and IgG binding was detected using a phycoerythrin (PE)-conjugated mouse anti-monkey IgG (Southern Biotech, Birmingham, Alabama) at 2 μg/ml. Plates were washed and acquired on a Bio-Plex 200 instrument (Bio-Rad, Hercules, CA), and IgG binding was reported as mean fluorescence intensity (MFI). To assess assay background, the MFIs of wells without sample (blank wells) were used, as well as evaluated nonspecific binding of the samples to unconjugated blank beads.
RBD-ACE2 Blocking Assay
Corning 384-well plates were coated with 3.5 ug/mL ACE2 protein (Sinobiological) 24 hours before conducting the experiment. The day of the assay, the plate was blocked with assay diluent (phosphate-buffered saline containing 4% whey, 15% normal goat serum, and 0.5% Tween 20) for 1 hour at room temperature. Plasma was diluted 1:10, 1:40, or 1:60 using the assay diluent, and incubated with 1 ug/mL HRP-RBD protein (Genescript) at 37C for 1 hour. Plates were washed and the preincubated mixture of plasma and HRP-RBD protein was added in duplicates and incubated at RT for 1 hour. Then, plates were washed 4 times to remove unbound sample, and peroxidase substrate solution (SeraCare) was added for 4 min before stopping the reaction using stop solution (SeraCare). Signal was detected using a Spectramax plate reader at OD 450. The amount of signal detected in wells without sample (diluent only) was considered the maximal binding response, and the OD450 detected in the sample wells was transformed to percentage inhibition compared to maximum binding. Data presented is the average of two replicates when CV is less than 20%, and if different assays were performed, median value was considered.
Pseudovirus Antibody Neutralization Assay
SARS-CoV-2 neutralization was assessed with Spike-pseudotyped viruses in 293T/ACE2 cells as a function of reductions in luciferase (Luc) reporter activity. 293T/ACE2 cells were kindly provided by Drs. Farzan and Mu at Scripps Florida. Cells were maintained in DMEM containing 10% FBS, 25 mM HEPES, 50 μg/ml gentamycin and 3 μg/ml puromycin. An expression plasmid encoding codon-optimized full-length Spike of the Wuhan-1 strain (VRC7480), was provided by Drs. Graham and Corbett at the Vaccine Research Center, National Institutes of Health (USA). The D614G amino acid change was introduced into VRC7480 by site-directed mutagenesis using the QuikChange Lightning Site-Directed Mutagenesis Kit from Agilent Technologies (Catalog # 210518). The mutation was confirmed by full-length Spike gene sequencing. Pseudovirions were produced in HEK 293T/17 cells (ATCC cat. no. CRL-11268) by transfection using Fugene 6 (Promega Cat#E2692) and a combination of Spike plasmid, lentiviral backbone plasmid (pCMV ΔR8.2) and firefly Luc reporter gene plasmid (pHR’ CMV Luc) (78) in a 1:17:17 ratio. Transfections were allowed to proceed for 16-20 hours at 37oC. Medium was removed, monolayers rinsed with growth medium, and 15 ml of fresh growth medium added. Pseudovirus-containing culture medium was collected after an additional 2 days of incubation and was clarified of cells by low-speed centrifugation and 0.45 μm micron filtration and stored in aliquots at -80°C. TCID50 assays were performed on thawed aliquots to determine the infectious dose for neutralization assays.
For neutralization, a pre-titrated dose of pseudovirus was incubated with 8 serial 5-fold dilutions of serum samples in duplicate in a total volume of 150 μl for 1 hour at 37oC in 96-well flat-bottom poly-L-lysine-coated culture plates (Corning Biocoat). Cells were suspended using TrypLE Select Enzyme solution (Thermo Fisher Scientific) and immediately added to all wells (10,000 cells in 100 μL of growth medium per well). One set of 8 control wells received cells + virus (virus control) and another set of 8 wells received cells only (background control). After 66-72 hours of incubation, medium was removed by gentle aspiration and 30 μL of Promega 1X lysis buffer was added to all wells. After a 10-min incubation at room temperature, 100 μl of Bright-Glo luciferase reagent was added to all wells. After 1-2 min, 110 μl of the cell lysate was transferred to a black/white plate (Perkin-Elmer). Luminescence was measured using a PerkinElmer Life Sciences, Model Victor2 luminometer. Neutralization titers are the serum dilution at which relative luminescence units (RLU) were reduced by either 50% (ID50) or 80% (ID80) compared to virus control wells after subtraction of background RLUs. Serum samples were heat-inactivated for 30 min at 56C prior to assay.
Whole Virus Neutralization Assay
Neutralization of SARS-CoV-2 nanoLUC carrying the D614G mutation was assessed as described in Hou et al. with modifications (79). Briefly, under BSL-3 containment, serially diluted sera at 8 dilutions were incubated for one hour with SARS-CoV-2 D614G nanoLUC virus at 5% CO2 and 37°C. After incubation, the virus/antibody mixtures were added in duplicate to black 96-well plates containing Vero E6 cells (2×104 cells/well). Each plate contains virus-only (no serum) control wells. The plates were incubated for 24 hours at 37°C, 5% CO2, the cells lysed, and luciferase activity measured with the Nano-Glo Luciferase Assay System (Promega). Neutralization activity is expressed as the dilution concentration at which the observed relative light units (RLU) are reduced by 50% or 80% relative to virus-only control wells.
Preparation of S-specific Hook Reagents for Flow Cytometry
To express the prefusion S ectodomain, a gene encoding residues 1−1208 of 2019-nCoV S (GenBank: MN908947) with proline substitutions at residues 986 and 987, a “GSAS” substitution at the furin cleavage site (residues 682–685), a C-terminal T4 fibritin trimerization motif, an HRV3C protease cleavage site, a TwinStrepTag and an 8XHisTag was synthesized and cloned into the mammalian expression vector pαH. To express the 2019-nCoV RBD-SD1, residues 319−591 of 2019-nCoV S were cloned upstream of a C-terminal HRV3C protease cleavage site, a monomeric Fc tag and an 8XHisTag. Similarly, to express the SARS-CoV RBD-SD1, residues 306−577 of SARS-CoV S (Tor2 strain) were cloned upstream of a C-terminal HRV3C protease cleavage site, a monomeric Fc tag and an 8XHisTag. Dimers are prepared based on the molar ratio (2:1) of the analyte protein and fluorochrome-conjugated Strep-Tactin, respectively. PE (IBA GmbH, 6-5000-001) or APC (IBA GmbH, 6-5010-001) conjugated Strep-Tactin was reacted with the protein over 5 additions, incubating for 15 min between each addition. The final concentration of tetramer was calculated with respect to the analyte protein. The solution was aliquoted based on the expected usage per experiment, snap frozen, and stored at -80°C. For quality control, monoclonal antibodies were bound to polystyrene beads (Spherotech) per the manufacturer’s instructions. B cell hooks were tested on beads coated with Ab026204 and CH65; mAb Ab026204 binds SARS-CoV2, mAb CH65 binds influenza hemagglutinin (80) and was used a negative control. Briefly, 0.5 μL of beads were diluted to 25 μL in PBS+0.02% NaN3. The bead mixture was added to 6 wells (25 μL per well) in a 96-well filter plate. B cell hooks were diluted to a final concentration of 40 μg/mL in 175 μL of PBS+0.02% NaN3, then four serial 2-fold dilutions were made down to a final concentration of 5 μg/mL. Test conjugate dilutions (25 μL per well) were added to each bead set, with one additional well of each bead set left as an unstained control. After 30-min incubation, the solution was vacuumed off and the beads washed 3 times with 150 μL PBS+0.02% NaN3. The washed beads were resuspended in 125 μL of PBS+0.02% NaN3, and the samples were run on a BD LSRII cytometer within 4 hours of preparation. Data were analyzed using Flow Jo software (Treestar) and plots of each dilution compared to the unstained control.
Antigen-specific B Cell Quantitation
S-specific B cells were assessed by flow cytometry as described (39) (Table S1). Freshly isolated or archived (week 6) PBMC or LNC (2×106 cells) were washed with phosphate buffered saline (PBS, Gibco) and pelleted by centrifugation at 500 × g for 7 min. Cells were resuspended in 150 μL 1% bovine serum albumin (BSA, Sigma-Aldrich) + 5 μM Chk2 inhibitor II in PBS and incubated 15 min at 4C in the dark, and subsequently washed with PBS + 1% BSA (7 min at 500 × g). The cell pellet was stained with antibodies prescribed in Table S2 for 30 min in the dark at 4C. Total CD20+ and/or memory CD27+ S-specific B cells were identified as double-positive for biotinylated S-2P protein labeled with avidin-APC or avidin-PE (Fig. S14). Germinal center (GC) B cells were analyzed by flow cytometry (Table S2) as detailed elsewhere (81). After staining, cells were washed as before and fixed with 1% paraformaldehyde before immediate acquisition on an LSRFortessa (BD) using BD FACSDiva v.8.0 and analyzed with FlowJo software v10.7.1 (Fig. S15).
B Cell ELISpot
Polyvinylidene fluoride membranes (Millipore) were activated with 70% ethanol for 1 min, subsequently coated with 1 μg SARS-CoV-2 Spike protein (2019-nCoV) per well and blocked for 2 hours with 2% milk and stored at 4C. Fresh or thawed cell preparations were washed and stimulated (106 cells/mL) with 1 μg/mL R848 (InvivoGen, San Diego, CA) and 10 ng/mL human IL‐2 (Miltenyi Biotec) for 72 hours in complete medium (cRPMI): RPMI‐1640 medium (Gibco) containing penicillin, streptomycin, L‐glutamine (Sigma‐Aldrich), and 10% heat‐inactivated FBS (Gibco). Stimulated cells (8 × 104/well) were incubated in triplicate wells of SARS-CoV-2 Spike protein-coated microtiter plates overnight at 37°C, washed with PBS+0.05% Tween 20, and incubated 1 hour with 1 μg/mL biotinylated affinity‐purified goat anti‐human IgG (Southern Biotech) at room temperature. Plates were washed with PBS+0.05% Tween 20, incubated with a 1:4000 dilution of avidin‐peroxidase (Southern Biotech) for 1 hour at room temperature, and developed using the BD AEC kit using 100 μL per well. Dried membranes were analyzed with an automated ELISpot Reader System (Autoimmun Diagnostika GmbH). Results are reported as the number of antibody‐secreting cells (ASC) per 106 MNCs.
T Cell Responses
Cryopreserved cells were thawed, or fresh preparations were washed and cultured in cRPMI (106/mL) with or without 2 μg/mL overlapping peptides spanning the length of SARS-CoV-2 spike protein (JPT Technologies) or DMSO vehicle together with co-stimulatory antibodies against CD28 and CD49d from BD. Naïve donor cells were included with each assay and, in addition to peptide pool or vehicle, were stimulated with 0.5x cell stimulation cocktail (eBiosciences) as a positive control. Cells were surface stained as outlined in Table S2 and then permeabilized with BD CytoFix/CytoPerm per manufacturer’s recommendations and stained with intracellular antibodies as prescribed in Table S2. Data were collected using an LSRFortessa and BD FACSDiva v8.0 and analyzed with FlowJo software v10.7.1 (TreeStar). Intracellular cytokine gates (Fig. S16) were Boolean gated and reported as single positive events unless otherwise noted.
In addition, we performed the activation-induced marker (AIM) assay (49, 82) on cryopreserved LNC. Cells were thawed, rested 3 h at 37°C with 5% CO2, resuspended in AIM V medium (Gibco), and transferred at 106 cells per well to a 24-well plate. Cells were cultured with vehicle DMSO (negative control) or with 0.5 μg/ml staphylococcal enterotoxin B (Toxin Technologies) for 20 hours at 37°C with 5% CO2. After stimulation, cells were stained with prescribed antibodies per Table S2 and acquired immediately on an LSRFortessa instrument running FACSDiva v8.0 software (BD Biosciences) and analyzed as described above (see Fig. S17).
Plasma Cytokine Measurements
IL-2, IL-4, IL-13, and IFN-γ in undiluted plasma samples were quantified by a custom 4-plex rhesus macaque Luminex assay (Thermofisher Scientific) using protocols established by the supplier. ELISA results are reported as the average concentration of duplicate wells extrapolated from a standard curve.
Statistical Analyses
Spearman’s rank correlations were estimated between pre-specified parameters at specific timepoints. Statistical analyses were performed using SAS version 9.4 (Cary, NC, USA). ID50 neutralization titer decline half-life was estimated based on random-effects regression models of decay with first-order kinetics (83, 84). Models were fit separately by vaccine group and data prior to the second vaccine dose (i.e., from weeks 0 and 4) were excluded. Bi-phasic decline was modeled using a linear spline with one knot (85), with different knots considered ranging from 8 to 18 weeks. For each vaccine group, the model with the knot at 18 weeks fit the best according to the Akaike Information Criterion.
REFERENCES AND NOTES
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
Hippich M, Holthaus L, Assfalg R, Zapardiel-Gonzalo J, Kapfelsperger H, Heigermoser M, Haupt F, Ewald DA, Welzhofer TC, Marcus BA, Heck S, Koelln A, Stock J, Voss F, Secchi M, Piemonti L, de la Rosa K, Protzer U, Boehmer M, Achenbach P, Lampasona V, Bonifacio E, Ziegler AG. A Public Health Antibody Screening Indicates a 6-Fold Higher SARS-CoV-2 Exposure Rate than Reported Cases in Children. Med (N Y). 2021;2(2):149-63 e4.
- ↵
Hippich M, Sifft P, Zapardiel-Gonzalo J, Bohmer MM, Lampasona V, Bonifacio E, Ziegler AG. A Public Health Antibody Screening Indicates a Marked Increase of SARS-CoV-2 Exposure Rate in Children during the Second Wave. Med (N Y). 2021. Epub 2021/04/13. doi: . PubMed PMID: 33842906; PMCID: PMC8018829.10.1016/j.medj.2021.03.019
- ↵
-
Loades ME, Chatburn E, Higson-Sweeney N, Reynolds S, Shafran R, Brigden A, Linney C, McManus MN, Borwick C, Crawley E. Rapid Systematic Review: The Impact of Social Isolation and Loneliness on the Mental Health of Children and Adolescents in the Context of COVID-19. J Am Acad Child Adolesc Psychiatry. 2020;59(11):1218-39 e3.
- ↵
- ↵
-
Mintz K, Jardas E, Shah S, Grady C, Danis M, Wendler D. Enrolling Minors in COVID-19 Vaccine Trials. Pediatrics. 2021;147(3). Epub 2020/12/19. doi: . PubMed PMID: 33334920; PMCID: PMC7919110 conflicts of interest to disclose. 10.1542/peds.2020-040717
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
Sekine T, Perez-Potti A, Rivera-Ballesteros O, Stralin K, Gorin JB, Olsson A, Llewellyn-Lacey S, Kamal H, Bogdanovic G, Muschiol S, Wullimann DJ, Kammann T, Emgard J, Parrot T, Folkesson E, Karolinska C-SG, Rooyackers O, Eriksson LI, Henter JI, Sonnerborg A, Allander T, Albert J, Nielsen M, Klingstrom J, Gredmark-Russ S, Bjorkstrom NK, Sandberg JK, Price DA, Ljunggren HG, Aleman S, Buggert M. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell. 2020;183(1):158-68 e14.
- ↵
- ↵
- ↵
-
Philbin VJ, Dowling DJ, Gallington LC, Cortes G, Tan Z, Suter EE, Chi KW, Shuckett A, Stoler-Barak L, Tomai M, Miller RL, Mansfield K, Levy O. Imidazoquinoline Toll-like receptor 8 agonists activate human newborn monocytes and dendritic cells through adenosine-refractory and caspase-1-dependent pathways. J Allergy Clin Immunol. 2012;130(1):195-204 e9.
- ↵
- ↵
- ↵
Data. OWi. Number of births and death per year, world University of Oxford2021 [cited 2021 May 28, 2021].
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- Copyright © 2021, American Association for the Advancement of Science