Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused a global pandemic, resulting millions of infections and deaths with few effective interventions available. Here, we demonstrate that SARS-CoV-2 evades interferon (IFN) activation in respiratory epithelial cells, resulting in a delayed response in bystander cells. Since pretreatment with IFNs can block viral infection, we reasoned that pharmacological activation of innate immune pathways could control SARS-CoV-2 infection. To identify potent antiviral innate immune agonists, we screened a panel of 75 microbial ligands that activate diverse signaling pathways and identified cyclic dinucleotides (CDNs), canonical STING agonists, as antiviral. Since CDNs have poor bioavailability, we tested the small molecule STING agonist diABZI, and found that it potently inhibits SARS-CoV-2 infection of diverse strains including variants of concern (B.1.351) by transiently stimulating IFN signaling. Importantly, diABZI restricts viral replication in primary human bronchial epithelial cells and in mice in vivo. Our study provides evidence that activation of STING may represent a promising therapeutic strategy to control SARS-CoV-2.
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of coronavirus disease 2019 (COVID-19), is a novel coronavirus that emerged in Wuhan, China in December 2019 (1). As a highly transmissible and pathogenic virus, SARS-CoV-2 rapidly spread around the world, causing a pandemic that poses a significant threat to global public health. As of January 2021, SARS-CoV-2 has infected more than 100 million people and resulted in more than 2 million deaths worldwide (2). Therefore, the development of effective antivirals is urgently needed for controlling SARS-CoV-2 infection and disease.
SARS-CoV-2 initially targets respiratory epithelial cells (1, 3). As the first line of defense against infection, the host innate immune system within the respiratory tract recognizes viral pathogens by detecting their pathogen-associated molecular patterns (PAMPs) through host-encoded pattern recognition receptors (PRRs) (4). Viral recognition by PRRs activates downstream signaling cascades, leading to the induction of effectors including type I and type III interferons (IFNs) (5). IFNs subsequently bind to their specific receptors in an autocrine and paracrine manner to activate JAK/STAT signaling and up-regulate the expression of hundreds of interferon-stimulated genes (ISGs), conferring an antiviral state to the host cells. Therefore, activation of IFN and other innate immune pathways can potentially block infection. Indeed, there are clinical trials underway to treat COVID-19 with IFNs with mixed success (6, 7)
We set out to characterize the innate response to SARS-CoV-2 and found that there was delayed activation due to immune evasion. This suggested that there may be additional innate immune agonists that could block SARS-CoV-2 infection, and perhaps small molecules with drug-like properties would have advantages over the protein IFN. To identify antiviral innate immune agonists to block viral infection, we performed high throughput screening and identified endogenous STING agonists, cyclic dinucleotides (CDNs), as antiviral against SARS-CoV-2. STING activation leads to the induction of IFNs as well as inflammatory NF-κB signaling and can inhibit diverse viral infections (8). While CDNs have low potency and make poor drugs, potent small molecule STING agonists have been developed including diABZI (9). We found that diABZI was active against SARS-CoV-2 in primary human respiratory epithelial cells and in vivo in two different mouse models of infection. We also found that we could treat infection therapeutically against diverse strains of SARS-CoV-2 suggesting that this STING agonist may serve as a novel therapeutic strategy to combat COVID-19.
RESULTS
SARS-CoV-2 induces immune activation at late time points in human respiratory epithelial cells
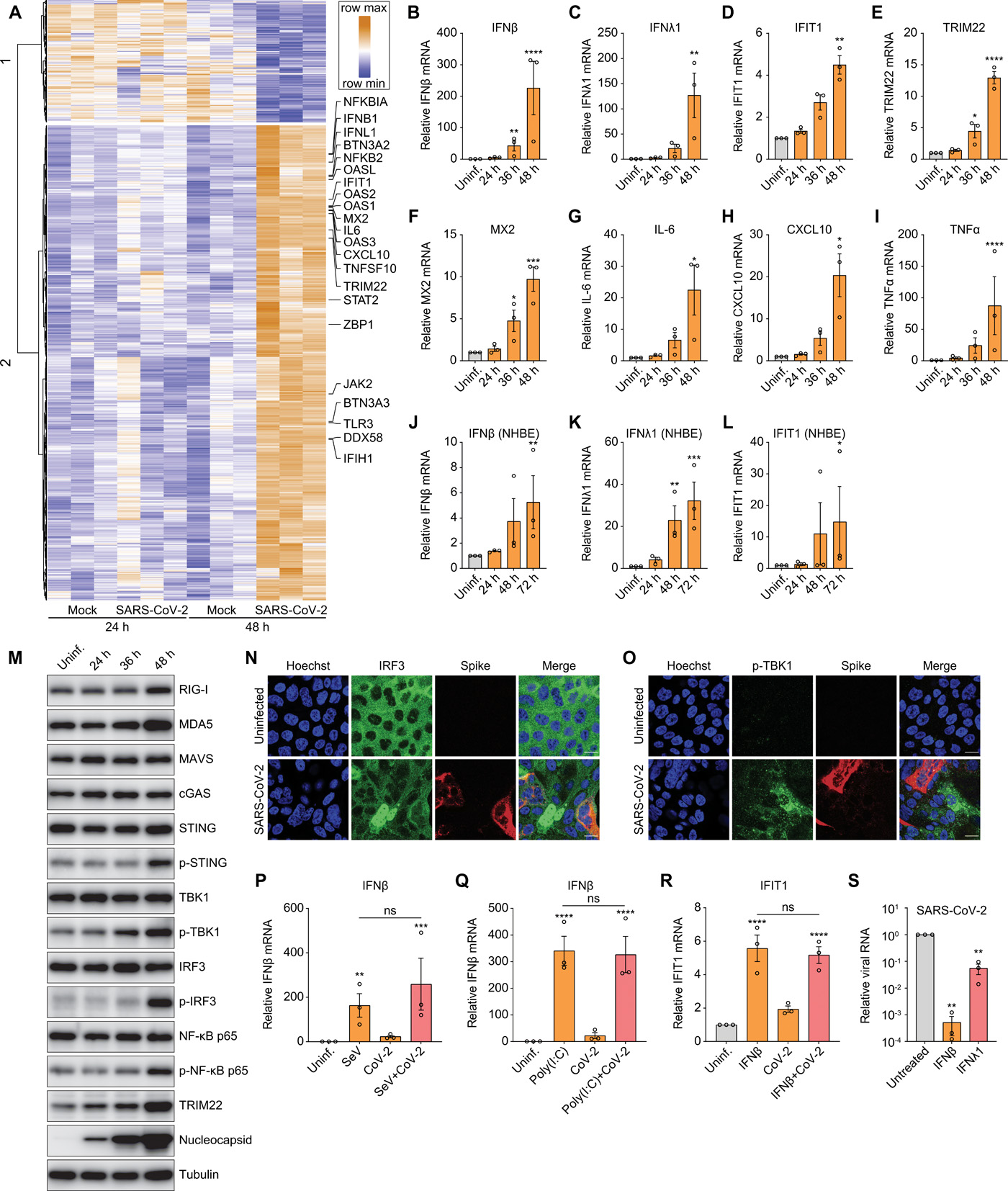
Although replication of SARS-CoV-2 in the respiratory tract stimulates innate immunity, the responses are attenuated compared to those observed after infection with other respiratory viruses (1, 10). Indeed, using RNAseq, we found that human lung epithelial Calu-3 cells show few transcriptional changes at 24 hours post-infection (hpi), whereas at 48 hpi 988 genes are induced (adjusted p 1, avg reads > 20 across all samples). Gene ontology (GO) enrichment analysis reveals that genes associated with IFN and antiviral defense are induced at the 48 hours time point (Fig. 1A and S1A). We validated the induction of type I (IFNβ) and III (IFNλ1) IFNs, as well as ISGs (e.g., IFIT1, TRIM22, and MX2) during infection by RT-qPCR (Fig. 1B to F). In addition, we validated that proinflammatory cytokines and chemokines (e.g., IL-6, CXCL10 and TNFα) are induced in SARS-CoV-2-infected Calu-3 cells by RT-qPCR (Fig. 1G to I). We also observed that SARS-CoV-2 induced type I IFNs, type III IFNs and ISG expression in primary normal human bronchial epithelial (NHBE) cells at late time points post infection (Fig. 1J to L).
(A) Calu-3 cells were mock-infected or infected with SARS-CoV-2 for 24 or 48 hours in 3 independent experiments. Total RNA was extracted and subjected to RNAseq analysis. Heat map shows the genes with greater than 1 log2 fold change, an adjusted P value less than 0.05, and average read number greater than 20. (B to I) Calu-3 cells were either uninfected (Uninf.) or infected with SARS-CoV-2 (MOI of 0.5) for 24, 36, or 48 hours. mRNA expression of IFNβ (B), IFNλ1 (C), IFIT1 (D), TRIM22 (E), MX2 (F), IL-6 (G), CXCL10 (H), and TNFα (I) was examined by RT-qPCR. Gene expression (gene/18S) was normalized to uninfected cells. (J to L) NHBE cells were either uninfected (Uninf.) or infected with SARS-CoV-2 (MOI of 0.5) for 24, 48 or 72 hours. mRNA expression of IFNβ (J), IFNλ1 (K) and IFIT1 (L) was quantified by RT-qPCR. Gene expression (gene/18S) was normalized to uninfected cells. (M) Calu-3 cells were uninfected (Uninf.) or infected with SARS-CoV-2 (MOI of 0.5) for the indicated time points. Cells were lysed, and the protein expression level was determined by immunoblotting using indicated antibodies. Representative blots of 3 independent experiments are shown. (N and O) Calu-3 cells were uninfected or infected with SARS-CoV-2 (MOI of 0.5) for 48 hours. Cells were fixed and stained with antibodies against IRF3 (N) or p-TBK1 (O). Representative images of 3 independent experiments are shown. Scale bar: 20 μm. (P and Q) Calu-3 cells were uninfected (Uninf.) or infected with SARS-CoV-2 (MOI of 0.5) for 24 hours. The uninfected or infected cells were then treated with Sendai virus (SeV) for 8 hours (P) or poly(I:C) for 6 hours (Q). The levels of IFNβ mRNA were quantified by RT-qPCR. (R) Calu-3 cells were uninfected (Uninf.) or infected with SARS-CoV-2 (MOI of 0.5) for 24 hours. The uninfected or infected cells were treated with IFNβ for 8 hours. The expression of IFIT1 were analyzed by RT-qPCR. (S) Calu-3 cells were untreated or pretreated with 10 ng/mL of IFNβ or IFNλ1 for 1 hour and inoculated with SARS-CoV-2 (MOI of 0.2) for 48 hours. Viral replication was quantified by RT-qPCR. For all graphs, shown is the mean ± SEM for 3 independent experiments. The significance was calculated using one-way ANOVA and is indicated by (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant).
” data-icon-position=”” data-hide-link-title=”0″>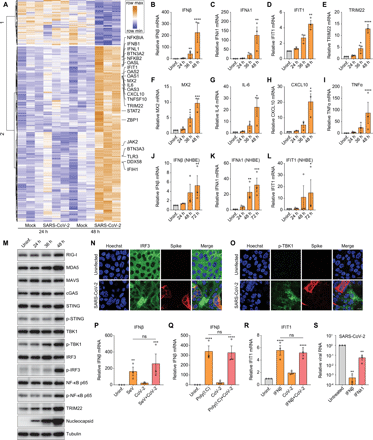
(A) Calu-3 cells were mock-infected or infected with SARS-CoV-2 for 24 or 48 hours in 3 independent experiments. Total RNA was extracted and subjected to RNAseq analysis. Heat map shows the genes with greater than 1 log2 fold change, an adjusted P value less than 0.05, and average read number greater than 20. (B to I) Calu-3 cells were either uninfected (Uninf.) or infected with SARS-CoV-2 (MOI of 0.5) for 24, 36, or 48 hours. mRNA expression of IFNβ (B), IFNλ1 (C), IFIT1 (D), TRIM22 (E), MX2 (F), IL-6 (G), CXCL10 (H), and TNFα (I) was examined by RT-qPCR. Gene expression (gene/18S) was normalized to uninfected cells. (J to L) NHBE cells were either uninfected (Uninf.) or infected with SARS-CoV-2 (MOI of 0.5) for 24, 48 or 72 hours. mRNA expression of IFNβ (J), IFNλ1 (K) and IFIT1 (L) was quantified by RT-qPCR. Gene expression (gene/18S) was normalized to uninfected cells. (M) Calu-3 cells were uninfected (Uninf.) or infected with SARS-CoV-2 (MOI of 0.5) for the indicated time points. Cells were lysed, and the protein expression level was determined by immunoblotting using indicated antibodies. Representative blots of 3 independent experiments are shown. (N and O) Calu-3 cells were uninfected or infected with SARS-CoV-2 (MOI of 0.5) for 48 hours. Cells were fixed and stained with antibodies against IRF3 (N) or p-TBK1 (O). Representative images of 3 independent experiments are shown. Scale bar: 20 μm. (P and Q) Calu-3 cells were uninfected (Uninf.) or infected with SARS-CoV-2 (MOI of 0.5) for 24 hours. The uninfected or infected cells were then treated with Sendai virus (SeV) for 8 hours (P) or poly(I:C) for 6 hours (Q). The levels of IFNβ mRNA were quantified by RT-qPCR. (R) Calu-3 cells were uninfected (Uninf.) or infected with SARS-CoV-2 (MOI of 0.5) for 24 hours. The uninfected or infected cells were treated with IFNβ for 8 hours. The expression of IFIT1 were analyzed by RT-qPCR. (S) Calu-3 cells were untreated or pretreated with 10 ng/mL of IFNβ or IFNλ1 for 1 hour and inoculated with SARS-CoV-2 (MOI of 0.2) for 48 hours. Viral replication was quantified by RT-qPCR. For all graphs, shown is the mean ± SEM for 3 independent experiments. The significance was calculated using one-way ANOVA and is indicated by (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant).
Since induction of ISGs was apparent at 48 but not 24 hpi in Calu-3 cells, we hypothesized there might be early evasion of canonical innate immune pathways. To evaluate this, we examined the activation status of diverse PRR and innate signaling pathways in Calu-3 cells. We monitored the canonical cytoplasmic viral RNA sensing pathway including the RIG-I/MDA5 receptors (RLRs), the adapter MAVS, and downstream signaling molecules TBK1 and IRF3 (Fig. 1M). We found that SARS-CoV-2 infection has a modest stimulatory effect on the expression of RIG-I, MDA5 and MAVS, but phosphorylation of TBK1 and IRF3 is more strongly induced, but again only at late time points after infection (Fig. 1M). Consistent with late activation of IRF3, the interferon-stimulated gene (ISG) TRIM22 is also induced at 48 hpi (Fig. 1M).
We also examined the expression of the DNA sensor cGAS that produces cyclic dinucleotides that activate STING (11–13). Although the cGAS-STING pathway has a key role in immune recognition of DNA viruses, it also has been found to play roles in RNA virus infections (14). SARS-CoV-2 infection leads to mild activation of STING as measured by phosphorylated STING at later stages of infection, but no change in cGAS was observed (Fig. 1M). Furthermore, STING does not have constitutive antiviral activity in Calu-3 cells, as treatment with the STING covalent antagonist H-151 did not affect infection as measured by microscopy or RT-qPCR (Fig. S1C and D) (15).
Inflammatory NF-κB signaling also was induced at 48 hpi, as measured by increased phospho-p65, which is consistent with the up-regulation of canonical targets, including TNFα (Fig. 1I and M). Taken together, these data suggest that SARS-CoV-2 infection elicits a delayed immune response in respiratory epithelial cells.
SARS-CoV-2 evades IFN activation but can be controlled by IFN treatment
To further explore how SARS-CoV-2 infection impacts innate immune signaling we monitored SARS-CoV-2-dependent activation of IRF3 and TBK1 at the single cell level using confocal microscopy. We observed virus-induced activation and nuclear accumulation of IRF3 at 48 hpi, but this was principally in uninfected bystander cells (Fig. 1N). Similarly, the up-regulation of phosphorylated TBK1 at 48 hpi occurred primarily in uninfected cells (Fig. 1O). To determine if this delayed response was due to delayed replication kinetics, we also monitored viral infection using an antibody to the viral replication intermediate dsRNA, which is also a potent immune stimulus, at an early time point (24 hpi). We detected robust viral dsRNA accumulation without apparent IRF3 nuclear translocation, further suggesting virus evades early immune detection (Fig. S1B). We confirmed that bystander activation of innate signaling was not specific to Calu-3 cells. We also observed virus-induced IRF3 translocation of bystander cells in the human respiratory cell line A549 that ectopically expresses ACE2 (Fig. S1E).
The lack of induction of ISGs early in infection could be due to viral antagonism of innate signaling pathways (16). We tested this hypothesis by inducing innate signaling using canonical ligands during SARS-CoV-2 infection. We included two stimuli that are known to induce IFNβ through RLR signaling: Sendai virus (SeV), and transfected poly(I:C). If SARS-CoV-2 antagonizes RLR signaling then we expect that the SeV or poly(I:C) induced IFN induction will be reduced in SARS-CoV-2-infected cells. Therefore, Calu-3 cells were either uninfected or infected with SARS-CoV-2 for 24 hours and then either co-infected with SeV (Fig. 1P) or transfected with poly(I:C) (Fig. 1Q). We observed similar levels of IFNβ induction by SeV or poly(I:C) in uninfected and co-infected cells; thus, SARS-CoV-2 does not antagonize RLR-dependent IFNβ mRNA induction. We also tested whether SARS-CoV-2 antagonizes IFN signaling and downstream ISG production. For these studies we treated cells with IFNβ and monitored IFNβ-induced up-regulation of ISGs IFIT1 and TRIM22. We found that the IFNβ-induced levels of ISG mRNAs were similar in uninfected and SARS-CoV-2-infected cells (Fig. 1R and S1F). These data suggest that SARS-CoV-2 is evading but not antagonizing PRR detection.
Next, we monitored this evasion using confocal microscopy which allowed us to assess the activation status of IRF3 in infected cells and uninfected bystander cells. We found that IRF3 nuclear translocation induced by SeV occurs in both uninfected and SARS-CoV-2-infected cells (Fig. S1G). This demonstrates that SeV-induced IRF3 activation is not antagonized by SARS-CoV-2. Again, we confirmed that this was not specific for Calu-3 cells as we observed that SeV induced IRF3 translocation in SARS-CoV-2 infected A549-ACE2 cells (Fig. S1H). Altogether, these data suggest that SARS-CoV-2 evades early recognition by canonical innate immune pathways.
Since SARS-CoV-2 was evading recognition, this suggested that treatment with interferons would induce antiviral gene expression and block SARS-CoV-2 infection. Whereas virtually all cells respond to type I IFNs, epithelial cells also respond to type III IFNs (17, 18). Therefore, we tested whether pretreatment of Calu-3 cells with different concentrations of type I IFNβ, or type III IFNλ1 would impact SARS-CoV-2 infection. We monitored both the percentage of infected cells and cell viability at 48 hpi using automated microscopy. Consistent with other studies (19, 20), IFNβ potently blocked SARS-CoV-2 infection at low concentration with an EC50 < 0.03 ng/mL and no apparent toxicity (Fig. S1, I and K). IFNλ1 is less active than IFNβ, and never fully blocked infection, even at high concentrations (Fig. S1, J and K). We confirmed the stronger antiviral activity of IFNβ than IFNλ1 by RT-qPCR (Fig. 1S). Since proteins such as IFNs are difficult to produce at scale for therapeutic use, we set out to identify additional drug-like molecules that could activate innate antiviral immunity.
High throughput screening identified STING agonists as antivirals against SARS-CoV-2
Indeed, we reasoned that additional innate immune agonists or pathogen-associated molecular patterns (PAMPs) could be used to suppress viral replication in respiratory cells. We screened a library of 75 agonists targeting diverse PRR sensing pathways in Calu-3 cells. These agonists include Toll-like receptor (TLR) agonists, RIG-I-like receptor (RLR) agonists, NOD-like receptor agonists, C-type lectin-like receptor agonists and cyclic dinucleotides (CDNs) (Table S1). We examined their effects on viral infection using automated microscopy and identified nine candidates that suppressed infection more than 10-fold (Fig. S2A). This included two CDNs (2’2′-cGAMP and 2’3′-cGAMP), four TLR7 agonists (imiquimod, gardiquimod, loxoribine and adilipoline) and HMW poly(I:C) (Fig. 2A, S2A, and Table S1). We performed dose-response studies with eight of the nine agonists and found that only the CDNs that activate STING (2’2′-cGAMP and 2’3′-cGAMP) and HMW poly(I:C) were active (Fig. 2B, S2A to I). We confirmed the potency of the canonical STING ligand produced by cGAS 2’3′-cGAMP using RT-qPCR, which showed a greater than 100-fold reduction in viral RNA levels (Fig. 2C).
(A) Calu-3 cells were treated with 75 innate immune agonists targeting different PRR sensing pathways for 2 hours, followed by inoculation with SARS-CoV-2 (MOI of 0.5). At 48 hpi, cells were fixed and stained for dsRNA and nuclei, followed by imaging with an automated microscope. Z-scores were calculated to quantify viral infection relative to vehicle controls. The two CDNs are labeled in red. (B) Dose-response analysis of Calu-3 cells treated with 2’,3′-cGAMP at the indicated concentrations and infected with SARS-CoV-2 (MOI of 0.5) for 48 hours. Viral infection and cell viability were quantified and normalized to vehicle control. (C) Untreated or 2’,3′-cGAMP-treated (100 μg/mL) Calu-3 cells were inoculated with SARS-CoV-2 (MOI of 0.2) for 48 hours. Viral RNA was monitored by RT-qPCR. Shown is the mean ± SEM for 3 independent experiments. Significance was calculated using an unpaired, two-tailed Student’s t test (*P < 0.05). (D) Calu-3 cells treated with serial dilutions of diABZI were infected with SARS-CoV-2. At 48 hpi, cells were stained with antibody against dsRNA and nuclei, and imaged. Percentage of infection and cell viability relative to vehicle control are shown. EC50, CC50 and SI were quantified. (E) Calu-3 cells were pretreated with DMSO or 10 μM diABZI for 1 hour, followed by inoculation with SARS-CoV-2 (MOI of 0.2) for 48 hours. Viral RNA was quantified using RT-qPCR. Shown is the mean ± SEM for 3 independent experiments. Significance was calculated using an unpaired, two-tailed Student’s t test (**P < 0.01). (F) Calu-3 cells were uninfected (Uninf.) or infected with SARS-CoV-2 (MOI of 0.5) for 24 hours. The uninfected or infected cells were treated with diABZI for 6 hours. The induction of IFIT1 were analyzed by RT-qPCR. Shown is the mean ± SEM for 3 independent experiments. Significance was calculated using one-way ANOVA (*P < 0.05; ns, not significant). (G) Calu-3 cells were treated either with DMSO or 10 μM diABZI for 2, 4, or 6 hours. Immunoblotting was performed to assess the protein expression levels with the indicated antibodies. Representative blots of 3 independent experiments are shown. (H and I) Calu-3 cells were treated with DMSO or 10 μM diABZI for 0.5 hours. Cells were fixed and stained with antibodies against p-STING (H) or IRF3 (I). Images shown are representative of 3 independent experiments. Scale bar: 20 μm.
” data-icon-position=”” data-hide-link-title=”0″>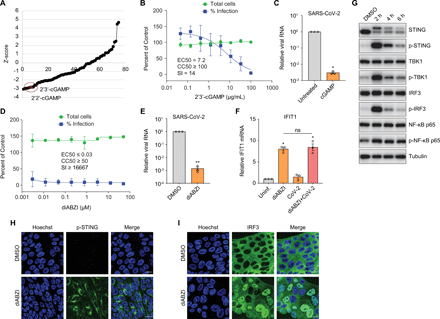
(A) Calu-3 cells were treated with 75 innate immune agonists targeting different PRR sensing pathways for 2 hours, followed by inoculation with SARS-CoV-2 (MOI of 0.5). At 48 hpi, cells were fixed and stained for dsRNA and nuclei, followed by imaging with an automated microscope. Z-scores were calculated to quantify viral infection relative to vehicle controls. The two CDNs are labeled in red. (B) Dose-response analysis of Calu-3 cells treated with 2’,3′-cGAMP at the indicated concentrations and infected with SARS-CoV-2 (MOI of 0.5) for 48 hours. Viral infection and cell viability were quantified and normalized to vehicle control. (C) Untreated or 2’,3′-cGAMP-treated (100 μg/mL) Calu-3 cells were inoculated with SARS-CoV-2 (MOI of 0.2) for 48 hours. Viral RNA was monitored by RT-qPCR. Shown is the mean ± SEM for 3 independent experiments. Significance was calculated using an unpaired, two-tailed Student’s t test (*P < 0.05). (D) Calu-3 cells treated with serial dilutions of diABZI were infected with SARS-CoV-2. At 48 hpi, cells were stained with antibody against dsRNA and nuclei, and imaged. Percentage of infection and cell viability relative to vehicle control are shown. EC50, CC50 and SI were quantified. (E) Calu-3 cells were pretreated with DMSO or 10 μM diABZI for 1 hour, followed by inoculation with SARS-CoV-2 (MOI of 0.2) for 48 hours. Viral RNA was quantified using RT-qPCR. Shown is the mean ± SEM for 3 independent experiments. Significance was calculated using an unpaired, two-tailed Student’s t test (**P < 0.01). (F) Calu-3 cells were uninfected (Uninf.) or infected with SARS-CoV-2 (MOI of 0.5) for 24 hours. The uninfected or infected cells were treated with diABZI for 6 hours. The induction of IFIT1 were analyzed by RT-qPCR. Shown is the mean ± SEM for 3 independent experiments. Significance was calculated using one-way ANOVA (*P < 0.05; ns, not significant). (G) Calu-3 cells were treated either with DMSO or 10 μM diABZI for 2, 4, or 6 hours. Immunoblotting was performed to assess the protein expression levels with the indicated antibodies. Representative blots of 3 independent experiments are shown. (H and I) Calu-3 cells were treated with DMSO or 10 μM diABZI for 0.5 hours. Cells were fixed and stained with antibodies against p-STING (H) or IRF3 (I). Images shown are representative of 3 independent experiments. Scale bar: 20 μm.
CDNs are negatively charged molecules that do not readily accumulate in the cell cytoplasm; therefore, CDNs are less potent in activating STING than newly developed small molecule agonists (21). Because of increased potency and drug-like properties, we next tested the activity of a non-nucleotide STING agonist diABZI on SARS-CoV-2 infection (Fig. S2J) (9). The dose-response analysis showed a dramatic inhibition of SARS-CoV-2 infection in diABZI-treated cells with a nanomolar EC50 and low toxicity (Fig. 2D). diABZI treatment led to a ~1,000-fold reduction in viral RNA levels as measured by RT-qPCR (Fig. 2E). We hypothesized that diABZI inhibits infection post-entry and indeed found that when diABZI was added to Calu-3 cells either 1 hour prior to infection (T = -1 hour) or 3 hours post infection (T = 3 hours) which is post entry, we observed a similar level of antiviral activity (Fig. S2K). Since we found that STING activation blocks infection, we also tested if SARS-CoV-2 antagonizes STING signaling. Compared to diABZI-treated uninfected cells, diABZI-dependent up-regulation of IFIT1 and IFNλ1 was similar in uninfected and SARS-CoV-2-infected cells (Fig. 2F and S2L). Therefore, SARS-CoV-2 does not antagonize STING signaling.
We verified that treatment with diABZI results in activation of STING in respiratory epithelial cells. A 2-hours treatment with diABZI results in rapid accumulation of a higher molecular weight species consistent with activation and phosphorylation, which we confirmed by using antibodies that recognize phosphorylated forms of STING (Fig. 2G). Phosphorylation of STING (Ser366) leads to both activation and subsequent degradation (22). Indeed, we observed the reduced expression of phosphorylated STING at 4 and 6 hours after diABZI treatment along with reductions in the higher molecular weight species and overall levels of STING over time (Fig. 2G). STING is known to activate TBK1, IRF3, and NF-κB signaling. Indeed, we detected increased phosphorylation of TBK1 and IRF3 at 2 hours post treatment that wanes over time closely mirroring the activation status of STING. Moreover, we found modest increases in NF-κB signaling as measured by phospho-p65 at 2 hours (Fig. 2G). We confirmed activation of STING by diABZI using confocal microscopy. STING activation and phosphorylation led to perinuclear accumulation and we observed phosphorylation and accumulation at perinuclear foci 30 min after diABZI treatment (Fig. 2H) (23). In addition, we observed diABZI-dependent activation of IRF3 as measured by its nuclear accumulation (Fig. 2I). Collectively, these data suggest diABZI activates the STING-TBK1-IRF3 signaling pathway in human respiratory Calu-3 epithelial cells.
STING activation potently and transiently induces innate signaling pathways
Activation of STING and its downstream signaling pathways induce antiviral gene programs that control infection (24). To determine the genes induced by diABZI, we performed RNAseq on Calu-3 cells treated for 6 or 12 hours. diABZI treatment leads to the robust activation of 403 genes, of which more than 146 are associated with antiviral IFN pathways (Fig. 3A and S3A). Indeed, we observed a remarkable induction of type I and III IFNs and ISGs after diABZI treatment (Fig. 3, B to H). Many classical ISGs including genes encoding OAS family proteins (OAS1, OAS2, OAS3, OASL) that are known human susceptibility/resistance loci are up-regulated in diABZI-treated cells (Fig. 3, I to K and S3B) (25, 26). diABZI treatment also causes a mild induction of NF-κB target gene TNFα (Fig. S3C).
(A) Calu-3 cells were treated with DMSO or 10 μM diABZI for 6 or 12 hours in two independent experiments. Total RNA was isolated and prepared for RNAseq analysis. Heatmap shows the differentially expressed genes with greater than 1 log2 fold change, an adjusted P value less than 0.05, and average read number greater than 20. (B to K) Calu-3 cells were treated with DMSO or 10 μM diABZI for the indicated time points. Total RNA was extracted and the induction of IFNβ (B), IFNλ1 (C), IFIT1 (D), TRIM22 (E), MX2 (F), IL6 (G), CXCL10 (H), OAS1 (I), OAS2 (J), and OAS3 (K) was determined using RT-qPCR. Shown is the mean ± SEM for 3 independent experiments. Asterisks indicate statistical significance as compared with DMSO-treated cells. (L to N) Calu-3 cells were incubated with 10 μM diABZI, 10 ng/mL IFNβ or IFNλ1 for the indicated time points. RNA was extracted and the expression of IFIT1 (L) or TRIM22 (M) mRNA was analyzed by RT-qPCR. Gene expression at 0 hour was set to 1. Shown is the mean ± SEM for 3 independent experiments. (N) Expression of IFIT1 and TRIM22 proteins was examined by Western blotting compared to tubulin control. Representative blots were shown. For all graphs, significance was calculated using one-way ANOVA (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
” data-icon-position=”” data-hide-link-title=”0″>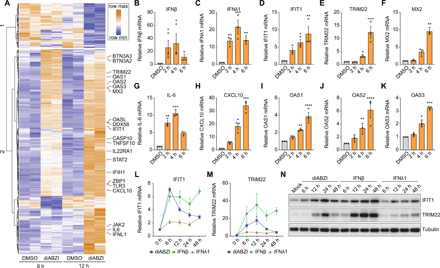
(A) Calu-3 cells were treated with DMSO or 10 μM diABZI for 6 or 12 hours in two independent experiments. Total RNA was isolated and prepared for RNAseq analysis. Heatmap shows the differentially expressed genes with greater than 1 log2 fold change, an adjusted P value less than 0.05, and average read number greater than 20. (B to K) Calu-3 cells were treated with DMSO or 10 μM diABZI for the indicated time points. Total RNA was extracted and the induction of IFNβ (B), IFNλ1 (C), IFIT1 (D), TRIM22 (E), MX2 (F), IL6 (G), CXCL10 (H), OAS1 (I), OAS2 (J), and OAS3 (K) was determined using RT-qPCR. Shown is the mean ± SEM for 3 independent experiments. Asterisks indicate statistical significance as compared with DMSO-treated cells. (L to N) Calu-3 cells were incubated with 10 μM diABZI, 10 ng/mL IFNβ or IFNλ1 for the indicated time points. RNA was extracted and the expression of IFIT1 (L) or TRIM22 (M) mRNA was analyzed by RT-qPCR. Gene expression at 0 hour was set to 1. Shown is the mean ± SEM for 3 independent experiments. (N) Expression of IFIT1 and TRIM22 proteins was examined by Western blotting compared to tubulin control. Representative blots were shown. For all graphs, significance was calculated using one-way ANOVA (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Moreover, we compared the kinetics of gene expression between IFNs and diABZI. Both diABZI and IFNβ induce ISGs (IFIT1, TRIM22, MX2) with maximal activation within 6 or 12 hours of treatment (Fig. 3, L and M, and S3D). However, IFNβ-induced ISGs expression is more sustained, whereas diABZI transiently up-regulates ISGs, followed by a more rapid decline. We confirmed the distinct kinetics of IFIT1 and TRIM22 by immunoblot where again both diABZI and IFNβ induce protein accumulation but the activation by diABZI is more transient (Fig. 3N). We also monitored expression of two NF-κB gene targets (CXCL10 and TNFα) and found transient activation by diABZI and IFNβ (Fig. S3, E and F). Consistent with its lack of antiviral activity, IFNλ1 has a minimal effect on the induction of ISGs or NF-κB targets that were profiled (Fig. 3, L and M, and S3, D to F).
STING activation blocks SARS-CoV-2 in human primary respiratory cells and in vivo
We next examined the antiviral activity of diABZI in primary normal human bronchial epithelial (NHBE) cells. NHBE cells were cultured either in submersion culture (NHBE-Submersion) or in an air-liquid interface (NHBE-ALI). Treatment with diABZI in NHBE-Submersion restricts SARS-CoV-2 replication, with a similar magnitude as remdesivir, an inhibitor of SARS-CoV-2 RNA replication (Fig. 4A). In the air-liquid interface culture system, which more closely mimics the human respiratory tract, both type I and III IFNs inhibit viral infection, although type III IFN is less active (Fig. 4D). Furthermore, treatment with diABZI blocked infection (~1,000-fold), similar to type I interferon treatment in these ALI cultures (Fig. 4, B to D, and S4).
(A) Primary normal human bronchial epithelial (NHBE) cells cultured in submersion culture (NHBE-Submersion) were treated with diABZI (10 μM) or remdesivir (10 μM), followed by infection of SARS-CoV-2 (MOI of 0.2). Viral RNA was quantified 72 hpi using RT-qPCR. Graphs show the mean ± SEM compared to DMSO-treated cells for 3 independent experiments. Significance was calculated using one-way ANOVA and is indicated by ***P < 0.001 as compared with DMSO-treated cells. (B and C) NHBE cells cultured in an air-liquid interface (NHBE-ALI) were mock-treated, or treated with DMSO or diABZI (10 μM) and infected with SARS-CoV-2 (MOI of 0.2). 72 hpi cells were then fixed and stained with SARS-CoV-2 Spike and nucleocapsid antibodies. Confocal microscopy was used to image the whole insert. Representative images are shown in (B) and quantified in (C) for two independent experiments. Significance was calculated using one-way ANOVA and is indicated by ***P < 0.001 as compared with DMSO control. (D) NHBE-ALI cells were pretreated with diABZI (10 μM), IFNβ (10 ng/mL) or IFNλ1 (10 ng/mL) and challenged with SARS-CoV-2 (MOI=0.2). RT-qPCR was used to quantify the level of viral RNA 72 hpi. Graphs show the mean ± SEM compared to DMSO-treated cells for 4 independent experiments. Significance was calculated using one-way ANOVA (****P < 0.0001) as compared with DMSO-treated cells. (E to G) Wild type C57BL/6 mice were treated with vehicle (PBS) or 10μg diABZI intranasally. At 6 or 12 hours post-treatment, lung tissues were collected and homogenized, followed by RNA extraction and RNAseq analysis. (E) Heatmap shows gene expression profile upon PBS or diABZI treatment of genes with a log2 fold change greater than 1 in at least one condition, an adjusted P value less than 0.05, and average read number greater than 20. (F and G) The expression of mouse Ifit1 was confirmed by RT-qPCR at 6 hours (F) or 12 hours (G) post-treatment (n = 5, Mann-Whitney test, is indicated by *P < 0.05; **P < 0.01). (H to K) K18-hACE2-transgenic mice were treated with vehicle control or 10μg diABZI via intranasal route 1 day prior (D-1) to infection and the day of infection (D0). Mice were inoculated via intranasal route with 103 PFU of SARS-CoV-2. (H) Body weight change of vehicle- or diABZI-treated mice was monitored daily (two independent experiments; from Day -1 to Day 3, n = 12 for Vehicle, n = 13 for diABZI; from Day 4 to Day 7, n = 8 for both Vehicle and diABZI; two-way ANOVA; P values: ***P = 0.0003 and ****P < 0.0001). At 3 and 7 dpi, viral burden in the lung tissues was determined by RT-qPCR for viral RNA (I and J) (n = 4 for Vehicle, n = 5 for diABZI; Mann-Whitney test; P value: *P = 0.0159 (I); n = 8; Mann-Whitney test; P value: ***P = 0.0002 (J)) (K) Viral load in the nasal wash was examined by RT-qPCR at the indicated time points (Day 3, n = 4 for Vehicle, n = 5 for diABZI; Day 7, n = 6 for Vehicle, n = 8 for diABZI; Mann-Whitney test; P values: **P = 0.0027 and *P = 0.0159). (L and M) K18-hACE2-transgenic mice were treated with vehicle control or 10 μg diABZI via intranasal route 1 day prior (D-1) to infection and the day of infection (D0) or 6 hours prior to infection (-6 hours). Survival rates (L) and body weight changes (M) of vehicle- or diABZI-treated mice were monitored daily (from Day -1 to Day 7, n = 8 for Vehicle (D-1/D0); from Day -1 to Day 8, n = 8 for Vehicle (-6h); from Day -1 to Day 14, n = 9 for diABZI (D-1/D0); from Day -1 to Day 14, n = 8 for diABZI (-6h). Significance in (L) was calculated using Log-rank (Mantel-Cox) test. P values: ***P = 0.0005; ****P < 0.0001. (N) K18-hACE2-transgenic mice were treated with vehicle control or indicated dose of diABZI at D0 and D-1 via intranasal route, followed by viral infection. Body weight change was monitored daily (n = 5 for both Vehicle and diABZI).
” data-icon-position=”” data-hide-link-title=”0″>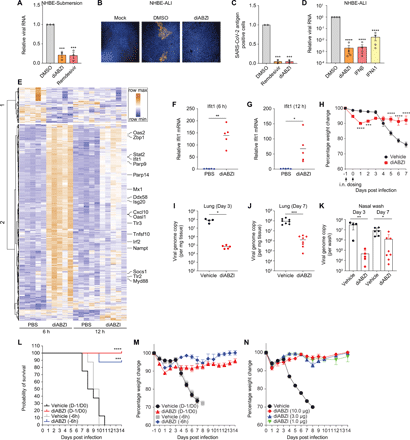
(A) Primary normal human bronchial epithelial (NHBE) cells cultured in submersion culture (NHBE-Submersion) were treated with diABZI (10 μM) or remdesivir (10 μM), followed by infection of SARS-CoV-2 (MOI of 0.2). Viral RNA was quantified 72 hpi using RT-qPCR. Graphs show the mean ± SEM compared to DMSO-treated cells for 3 independent experiments. Significance was calculated using one-way ANOVA and is indicated by ***P < 0.001 as compared with DMSO-treated cells. (B and C) NHBE cells cultured in an air-liquid interface (NHBE-ALI) were mock-treated, or treated with DMSO or diABZI (10 μM) and infected with SARS-CoV-2 (MOI of 0.2). 72 hpi cells were then fixed and stained with SARS-CoV-2 Spike and nucleocapsid antibodies. Confocal microscopy was used to image the whole insert. Representative images are shown in (B) and quantified in (C) for two independent experiments. Significance was calculated using one-way ANOVA and is indicated by ***P < 0.001 as compared with DMSO control. (D) NHBE-ALI cells were pretreated with diABZI (10 μM), IFNβ (10 ng/mL) or IFNλ1 (10 ng/mL) and challenged with SARS-CoV-2 (MOI=0.2). RT-qPCR was used to quantify the level of viral RNA 72 hpi. Graphs show the mean ± SEM compared to DMSO-treated cells for 4 independent experiments. Significance was calculated using one-way ANOVA (****P < 0.0001) as compared with DMSO-treated cells. (E to G) Wild type C57BL/6 mice were treated with vehicle (PBS) or 10μg diABZI intranasally. At 6 or 12 hours post-treatment, lung tissues were collected and homogenized, followed by RNA extraction and RNAseq analysis. (E) Heatmap shows gene expression profile upon PBS or diABZI treatment of genes with a log2 fold change greater than 1 in at least one condition, an adjusted P value less than 0.05, and average read number greater than 20. (F and G) The expression of mouse Ifit1 was confirmed by RT-qPCR at 6 hours (F) or 12 hours (G) post-treatment (n = 5, Mann-Whitney test, is indicated by *P < 0.05; **P < 0.01). (H to K) K18-hACE2-transgenic mice were treated with vehicle control or 10μg diABZI via intranasal route 1 day prior (D-1) to infection and the day of infection (D0). Mice were inoculated via intranasal route with 103 PFU of SARS-CoV-2. (H) Body weight change of vehicle- or diABZI-treated mice was monitored daily (two independent experiments; from Day -1 to Day 3, n = 12 for Vehicle, n = 13 for diABZI; from Day 4 to Day 7, n = 8 for both Vehicle and diABZI; two-way ANOVA; P values: ***P = 0.0003 and ****P < 0.0001). At 3 and 7 dpi, viral burden in the lung tissues was determined by RT-qPCR for viral RNA (I and J) (n = 4 for Vehicle, n = 5 for diABZI; Mann-Whitney test; P value: *P = 0.0159 (I); n = 8; Mann-Whitney test; P value: ***P = 0.0002 (J)) (K) Viral load in the nasal wash was examined by RT-qPCR at the indicated time points (Day 3, n = 4 for Vehicle, n = 5 for diABZI; Day 7, n = 6 for Vehicle, n = 8 for diABZI; Mann-Whitney test; P values: **P = 0.0027 and *P = 0.0159). (L and M) K18-hACE2-transgenic mice were treated with vehicle control or 10 μg diABZI via intranasal route 1 day prior (D-1) to infection and the day of infection (D0) or 6 hours prior to infection (-6 hours). Survival rates (L) and body weight changes (M) of vehicle- or diABZI-treated mice were monitored daily (from Day -1 to Day 7, n = 8 for Vehicle (D-1/D0); from Day -1 to Day 8, n = 8 for Vehicle (-6h); from Day -1 to Day 14, n = 9 for diABZI (D-1/D0); from Day -1 to Day 14, n = 8 for diABZI (-6h). Significance in (L) was calculated using Log-rank (Mantel-Cox) test. P values: ***P = 0.0005; ****P < 0.0001. (N) K18-hACE2-transgenic mice were treated with vehicle control or indicated dose of diABZI at D0 and D-1 via intranasal route, followed by viral infection. Body weight change was monitored daily (n = 5 for both Vehicle and diABZI).
To further explore the utility of STING agonists in vivo, we evaluated whether intranasal treatment of mice with diABZI induces an antiviral gene expression program. RNAseq analysis revealed a rapid activation of IFN signaling pathways in the lung after a single intranasal administration (Fig. 4, E to G, and S5, A to M). By comparing orthologs of genes induced by diABZI in Calu-3 human respiratory epithelial cells and mouse lungs, we found striking overlap suggesting a robust and common gene expression program across models (Fig. S6, A to D).
We next set out to determine if diABZI treatment in vivo was protective. K18 hACE2-transgenic mice express the human angiotensin-converting enzyme 2 (ACE2), a host cellular receptor for SARS-CoV-2 entry, and support high levels of SARS-CoV-2 infection in lung tissues, resulting in intense innate and adaptive immune cell infiltration and respiratory dysfunction (27). Using the K18-hACE2 transgenic mice, we evaluated the effect of diABZI on viral infection in vivo. Mice were pretreated with diABZI via intranasal route at day (D)-1, and given a second dose at the time of inoculation (D0) of SARS-CoV-2. At day 7 post-infection (dpi), whereas vehicle-treated mice had lost 25% of their body weight, diABZI-treated mice show much less weight loss (Fig. 4H). We did observe an early phase of weight loss (1 and 2 dpi) in diABZI-treated mice, which could reflect the induction of innate immune responses. Viral RNA levels in the lungs and nasal washes were reduced at 3 and 7 dpi (Fig. 4, I to K). We also quantified cytokine and chemokine proteins in mouse lung tissue at day 3 post infection comparing vehicle and diABZI treatment groups. As expected from our profiling studies, IFNs are induced by diABZI treatment as well as cytokines and chemokines involved in activation and recruitment of neutrophils (Fig. S5N). And there is specificity as a number of other cytokines and chemokines remain unchanged (Fig. S5O). These data further support that diABZI stimulates IFN for protective immunity.
Using the K18-hACE2 transgenic mice, we also evaluated whether diABZI can protect the animals from lethality and whether we could mitigate the weight loss observed upon diABZI treatment by reducing the dosing schedule to a single intranasal delivery of diABZI. Mice were pretreated with diABZI via intranasal route at day (D)-1, and given a second dose at the time of inoculation (D0) of SARS-CoV-2 or treated with one single dose at 6 hours prior to infection (-6 hours). Mice treated with diABZI with either regimen were protected from lethality (Fig. 4L). Moreover, animals treated only once had reduced weight loss early, and completely recovered at later time points (Fig. 4M). Since a single dose of 10 μg was protective, we also tested if we could reduce the dose of diABZI. We found that treatment with a single dose of 10 μg, 3 μg or 1 μg of diABZI delivered intranasally was protective from virus-induced weight loss and led to long-term survival (Fig. 4N). We also monitored lung histology comparing vehicle-treated mice to mice treated with a single 3 μg dose of diABZI delivered at the time of infection (Fig. S5P). Vehicle-treated infected lungs showed that immune cells, including neutrophils and mononuclear cells, infiltrated and accumulated throughout the lung in alveolar and interstitial locations with alveolar wall thickening. In contrast, mice treated with diABZI showed a marked attenuation of the inflammatory response in the lung with very few confined spots of cellular infiltration. Therefore, diABZI treatment decreases viral replication and lung inflammation.
The antiviral activity of STING activation is interferon-dependent
We next set out to explore the mechanism by which STING activation blocks SARS-CoV-2 infection. We treated cells with decreasing doses of diABZI, and found that there was ~1000-fold inhibition of SARS-CoV-2 replication at even the lowest dose tested (1 μM), and similar to the level of activity of high dose IFNβ (Fig. 5A). In addition, we tested the activity of both HMW poly(I:C) and LMW poly(I:C). We found that LMW poly(I:C) has little antiviral activity even at 100 μg/mL (Fig. 6A). In contrast, HMW poly(I:C) has strong antiviral activity at 100 μg/mL, but the activity drops off at 1 μg/mL (Fig. 5A).
(A and E) Calu-3 cells were untreated or pretreated with diABZI, IFNβ, poly(I:C)-LMW or poly(I:C)-HMW at indicated concentration for 1 hour and inoculated with SARS-CoV-2 USA-WA1/2020 strain (A) or South Africa B.1.351 strain (E) (MOI of 0.2) for 48 hours. Viral infection was then quantified by RT-qPCR. Graphs display the average fold change ± SEM compared to untreated cells in 3 independent experiments. (B and C) Calu-3 cells were pretreated with 10 μM diABZI alone, 100 μg/mL HMW poly(I:C) alone, 10 μM ruxolitinib alone or in combination for 1 hour, followed by SARS-CoV-2 infection (MOI of 0.2). At 48 hpi, viral RNA was examined by RT-qPCR and normalized to DMSO-treated (B) or untreated cells (C). Shown is the mean ± SEM for 3 independent experiments. (D) Calu-3 cells were treated with control siRNA or IFNAR1 siRNA for 2 days and then pretreated with 10 μM diABZI or 10 ng/mL IFNβ, followed by SARS-CoV-2 infection (MOI of 0.2). Viral RNA was examined by RT-qPCR at 48 hpi. Shown is the mean ± SEM for 3 independent experiments. (F) Calu-3 cells were treated with DMSO, 10 μM diABZI, 10 ng/mL IFNβ or 10 ng/mL IFNλ1 at 1 hour prior to SARS-CoV-2 (MOI of 0.2) infection (T = -1 hour), or 6 hours (T = 6 hours) or 12 hours (T = 12 hours) post-infection. At 48 hpi, viral replication was examined by RT-qPCR. Shown is the mean ± SEM for 3 independent experiments. For graphs in Fig. 5A to F, the significance was calculated using one-way ANOVA and is indicated by (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant). (G to I) Wild type 129 mice were treated with vehicle control or 3 μg diABZI via intranasal route 6 hours (-6 hours) prior to intranasal infection with 1×105 FFU of the SARS-CoV-2 Wash-SA B.1.351strain or 12 hours post-infection (+12 hours). (G) Body weight change was monitored daily (n =5 for both Vehicle and diABZI at -6 hours or +12 hours). Viral load in the lung (H) and nasal turbinate (I) was quantified at 4 dpi (n =5 for both Vehicle and diABZI; Mann-Whitney test; P value: **P = 0.0079 (H); *P = 0.0317, **P = 0.0079 (I)).
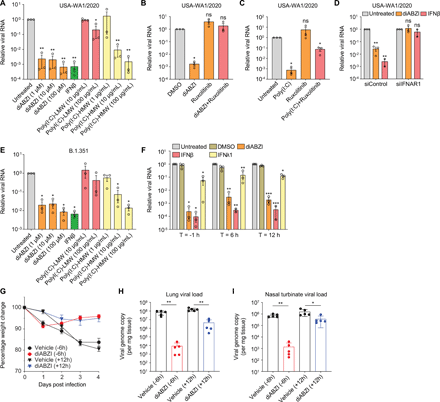
(A and E) Calu-3 cells were untreated or pretreated with diABZI, IFNβ, poly(I:C)-LMW or poly(I:C)-HMW at indicated concentration for 1 hour and inoculated with SARS-CoV-2 USA-WA1/2020 strain (A) or South Africa B.1.351 strain (E) (MOI of 0.2) for 48 hours. Viral infection was then quantified by RT-qPCR. Graphs display the average fold change ± SEM compared to untreated cells in 3 independent experiments. (B and C) Calu-3 cells were pretreated with 10 μM diABZI alone, 100 μg/mL HMW poly(I:C) alone, 10 μM ruxolitinib alone or in combination for 1 hour, followed by SARS-CoV-2 infection (MOI of 0.2). At 48 hpi, viral RNA was examined by RT-qPCR and normalized to DMSO-treated (B) or untreated cells (C). Shown is the mean ± SEM for 3 independent experiments. (D) Calu-3 cells were treated with control siRNA or IFNAR1 siRNA for 2 days and then pretreated with 10 μM diABZI or 10 ng/mL IFNβ, followed by SARS-CoV-2 infection (MOI of 0.2). Viral RNA was examined by RT-qPCR at 48 hpi. Shown is the mean ± SEM for 3 independent experiments. (F) Calu-3 cells were treated with DMSO, 10 μM diABZI, 10 ng/mL IFNβ or 10 ng/mL IFNλ1 at 1 hour prior to SARS-CoV-2 (MOI of 0.2) infection (T = -1 hour), or 6 hours (T = 6 hours) or 12 hours (T = 12 hours) post-infection. At 48 hpi, viral replication was examined by RT-qPCR. Shown is the mean ± SEM for 3 independent experiments. For graphs in Fig. 5A to F, the significance was calculated using one-way ANOVA and is indicated by (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant). (G to I) Wild type 129 mice were treated with vehicle control or 3 μg diABZI via intranasal route 6 hours (-6 hours) prior to intranasal infection with 1×105 FFU of the SARS-CoV-2 Wash-SA B.1.351strain or 12 hours post-infection (+12 hours). (G) Body weight change was monitored daily (n =5 for both Vehicle and diABZI at -6 hours or +12 hours). Viral load in the lung (H) and nasal turbinate (I) was quantified at 4 dpi (n =5 for both Vehicle and diABZI; Mann-Whitney test; P value: **P = 0.0079 (H); *P = 0.0317, **P = 0.0079 (I)).
STING activation is known to induce IFN expression, and we found that diABZI triggers IFN expression (Fig. 3A and 3B). Therefore, we tested whether IFN signaling was required for the diABZI-mediated antiviral response. For these studies, we used ruxolitinib, an inhibitor of JAK1/2, to block IFN-dependent gene expression (28). Calu-3 cells were pretreated with diABZI in the absence or presence of ruxolitinib, followed by SARS-CoV-2 infection for 48 hours. Whereas ruxolitinib alone had a mild effect on viral replication (Fig. 5B and S6E), ruxolitinib completely abrogated the diABZI-mediated restriction (Fig. 5B), suggesting that the antiviral activity of diABZI depends on JAK-STAT signaling. By contrast, ruxolitinib only partially blocked HMW poly(I:C)-mediated restriction, suggesting that in addition to IFN signaling, HMW poly(I:C) also stimulates alternative pathways to block viral infection (Fig. 5C). To further support the hypothesis that IFN signaling is required for diABZI function, we used a loss-of-function approach. We found that siRNA-mediated knockdown of IFNAR1 completely abolished diABZI antiviral activity (Fig. 5D).
STING activation restricts SARS-CoV-2 South African variant B.1.351
The emergence of SARS-CoV-2 variants has raised concern that some of these new strains may evade current vaccines (29). Therefore, there are increased efforts to identify treatments that would be active against these emerging variants. The South African variant B.1.351 is one such strain that has led to worldwide concern. Therefore, we tested the activity of diABZI, IFNβ, HMW poly(I:C) and LMW poly(I:C) against the South African variant B.1.351 (Fig. 5E). We found that there are similar trends with SARS-CoV-2 USA-WA1/202. diABZI has strong antiviral activity similar to IFNβ (Fig. 5E). LMW poly(I:C) shows little activity with HMW poly(I:C) having strong activity only at the highest dose (Fig. 5E). Altogether, diABZI showed robust antiviral activity against both strains of SARS-CoV-2.
While prophylactic dosing of diABZI was protective against SARS-CoV-2 (Fig. 2, Fig. 4, Fig. 5) we next explored if therapeutic dosing would also show activity. First, we tested this in Calu-3 cells. We treated cells with diABZI, IFNβ or IFNλ1 for 1 hour prior to infection, or 6 hours or 12 hours post-infection. Both diABZI and IFNβ potently restricted viral infection when pretreated or treated post-infection. In all cases, IFNλ1 was less active (Fig. 5A). This led us to test therapeutic dosing of diABZI in vivo.
For in vivo studies with the South African strain, the spike gene of B.1.351 was cloned into the WA1/2020 infectious clone (30) to produce Wash-B.1.351 chimeric virus, as described previously (31). This allowed us to infect wild type mice since the B.1.351 Spike can use murine ACE2 (32). Wild type 129 mice were treated with diABZI via the intranasal route at 6 hours (-6 hours) prior to infection or at 12 hours (+12 hours) post viral infection. diABZI treatment prior to infection or post infection both decreased viral load in mouse lung and nasal turbinate and protected mice from virus-induced weight loss (Fig. 5G to I). Moreover, there is no early weight loss in the animals treated with the single 3 μg dose (Fig. 5G). These data suggest that diABZI potently blocked infection of SARS-CoV-2 and can be used therapeutically.
DISCUSSION
In this study, we show that SARS-CoV-2 induces a delayed innate immune response in respiratory epithelial cells, which likely enables the virus to establish infection in the respiratory tract. The innate immune response that does develop appears restricted to bystander cells. Bystander cell activation is a common mechanism employed by immune system to alert neighboring uninfected cells of viral invasion via transfer of viral PAMPs or engagement of cytokines or metabolites produced by infected cells (33). However, this delayed response can be overcome by treatment with exogenous PAMPs or IFN, suggesting evasion but not antagonism by SARS-CoV-2.
This phenotype led us to hypothesize that additional innate immune pathways could block SARS-CoV-2 infection. Pharmacological activation of innate immune pathways has emerged as an attractive strategy for the treatment of cancer and viral diseases (34). Our screening of a library of 75 agonists targeting diverse innate sensing pathways identified STING agonists as potent inhibitors of SARS-CoV-2. Upon binding to its CDN ligand, STING activates the TBK1-IRF3 pathway to induce type I IFNs as well as proinflammatory cytokines (35). STING activation has been shown to be a promising strategy to treat cancers, leading to the development of small molecule agonists (9, 36). diABZI is a small-molecule, non-cyclic dinucleotide STING agonist that is more potent than endogenous CDNs (9). diABZI significantly suppresses SARS-CoV-2 infection in primary and transformed human respiratory epithelial cells, with marked reductions in infection that are comparable to type I IFN treatment. Moreover, we found that diABZI-mediated restriction of SARS-CoV-2 depends on IFN signaling. Furthermore, we found that prophylactic or therapeutic administration was protective in vivo.
There are few treatment options for COVID-19 with remdesivir and monoclonal antibodies as the only approved antivirals. These antivirals are typically restricted to use in a hospital setting; therefore, there is an urgent need for additional antivirals that may be given in an outpatient setting. Given the potency of IFNs in controlling viral infection, several clinical trials using type I IFN alone or in combination with other antiviral drugs are currently ongoing to treat patients infected with SARS-CoV-2 (37, 38). However, clinical trials with IFN treatment have had mixed results and many side effects (6, 7). Recent studies indicate that IFNs can damage lung epithelial barrier and impair lung epithelial regeneration during viral infection, highlighting the importance of timing of IFN administration for the treatment of lung viral diseases (39, 40). Therefore, the IFN response must be tightly regulated to avoid its adverse effects. We suggest that STING agonists may show promise. STING activation of IFN is quite transient (22), suggesting that early treatment could control infection with fewer complications. Indeed, we observed a transient induction of IFNs and ISGs in diABZI-treated cells and in mouse lungs, whereas IFNβ treatment induces sustained ISG expression. Moreover, we observed both decreased viral replication and inflammation in the lungs of mice treated with diABZI.
In addition, we found that therapeutic delivery of a single intranasal dose of diABZI was protective. Given the concerns arising with new variants of SARS-CoV-2, we hypothesized that diABZI would also control SARS-CoV-2 variants that may evade vaccines based on Spike recognition. Therefore, we tested the efficacy of diABZI against the South African variant B.1.351 and found that treatment is protective. This suggests that diABZI will be active against a broad array of SARS-CoV-2 variants and likely additional coronaviruses (41, 42). As diABZI also can inhibit human parainfluenza virus and rhinovirus replication in cultured cells (43), STING agonists may be more broadly active against respiratory viruses.
MATERIALS AND METHODS
Cells and viruses. Calu-3 cells (ATCC HTB-55) were cultured in Minimum Essential Medium (MEM) supplemented with 10% (v/v) fetal bovine serum, 1% (v/v) non-essential amino acids, 0.1% (v/v) penicillin/streptomycin, 4 mM L-glutamine at 37°C and 5% CO2. Plates and coverslips were coated with rat tail collagen (Corning) for 30 min and washed prior to plating. A549-ACE2 cells were cultured in RPMI-1640 1X, 10% FBS, 1% pen-strep (44). Normal human bronchial epithelial cells (NHBE, Lonza CC-2540) were cultured in suspension in Bronchial Epithelial Basal Medium (BEBM, Lonza CC-3171) supplemented with bovine pituitary extract, hydrocortisone, hEGF, epinephrine, transferrin, insulin, retinoic acid, triiodothyronine and gentamicin/amphotericin-B, according to the manufacturer’s recommendations and maintained at 37°C and 5% CO2. Cells were cultured for a maximum of three passages. Normal human bronchial epithelial cells were grown as ALI cultures (MatTek AIR-100) for >21 days post air exposure when robust beating cilia were observed. Cells are fed twice a week and on the day of infection with growth media AIR-100-MM (MatTek).
SARS-CoV-2 was obtained from BEI (USA WA1/2020 strain (A) or KRISP-EC-K005321/2020 (B.1.351)). Stocks were prepared by infection of Vero E6 cells in 2% serum plus 10 mM HEPES for five days, freeze-thawed, and clarified by centrifugation (P0). The titer of the stock was determined by plaque assay or tissue culture infectious dose using Vero E6 cells. This seed stock was sequence verified and amplified in Vero E6 (P1) and used for all experiments. All work with infectious virus was performed in a Biosafety Level 3 laboratory and approved by the Institutional Biosafety Committee and Environmental Health and Safety.
Agonist screen and dose-responses. A collection of 75 ligands recognized by pattern recognition receptors (PRRs) was purchased from InvivoGen (Table S1). Ligands were suspended in aqueous buffer and arrayed in 384-well plates as a 4-point dose response with 10-fold dilution between concentrations. Library plates were stored at -20°C and freeze-thawed a maximum of three times. Calu-3 cells (7,500 cells/well) were seeded in collagen coated 384-well plates (Corning BioCoat). The next day, 2.5 μL of the PRR library was added to the assay plates. The positive control (10 μM remdesivir, n=32) and the negative control (0.2% DMSO, n=32) were spotted on each assay plate. Two hours post addition of PRR agonist library, cells were inoculated with SARS-CoV-2 at a multiplicity of infection (MOI) of 0.5. Cells were fixed 48 hpi in 4% formaldehyde/PBS for 15 min at room temperature and then washed three times with PBS. Cells were blocked (2% BSA/PBST) for 60 min and incubated with anti-dsRNA antibody (mAbJ2) overnight at 4°C. Cells were washed 3x in PBST and incubated in secondary antibody (anti-mouse Alexa 488) and Hoechst 33342 for 1 hour at room temperature. Cells were washed 3x in PBST and imaged at 10X using ImagXpress Micro XLS (Molecular Devices) capturing four sites per well. The total number of cells and number of dsRNA positive cells were measured using the cell scoring module (MetaXpress 5.3.3), and the percentage of infected cells was calculated. Sample well infection was normalized to aggregated DMSO plate control wells and expressed as Percentage of Control [POC = (%Infectionsample/ Average %InfectionDMSO)*100] and Z-score [Z= (%Infectionsample – Average %InfectionDMSO) / Standard Deviation %InfectionDMSO] in Spotfire (PerkinElmer). Z-score is analogous to the standard deviation.
Dose-response studies: Drugs suspended in DMSO (or in aqueous solution for proteins or PAMPs), an 8-point dilution series with four-fold dilutions between test concentrations was prepared. Two hours prior to infection, 50 nL of drug in DMSO or 1 μL of aqueous solutions were added per well of assay plates, yielding the final drug concentration in 0.2% DMSO. Plates were infected as in the primary screen. Infection at each drug concentration was normalized to the aggregated DMSO control wells and expressed as percentage-of-control (POC=% Infection sample/Avg % Infection DMSO cont). A non-linear regression curve fit analysis (GraphPad Prism 8) was performed on the aggregated average POC Infection and cell viability from ≥ 2 independent experimental replicates versus the log10 transformed concentration values to calculate IC50 values for infection and CC50 values for cell viability. Error bars in dose response curves represent the standard deviation of replicate data for each drug concentration tested in independent experiments. Selectivity index (SI) was calculated as a ratio of drug’s CC50 and IC50 values (SI = CC50/IC50).
Reagents and antibodies. IFNβ and IFNλ1 were purchased from Biolegend. PAMPs were purchased from InvivoGen. diABZI, ruxolitinib and H-151 were purchased from Selleck Chemicals. The following antibodies were used for Western blotting or immunofluorescence: anti-RIG-I (Cell Signaling Technology, 3743S), anti-MDA5 (Cell Signaling Technology, 5321S), anti-MAVS (Cell Signaling Technology, 3993S), anti-cGAS (Cell Signaling Technology, 83623S), anti-STING (Cell Signaling Technology, 13647S), anti-STING (Proteintech, 19851-1-AP), anti-Phospho-STING (Ser366) (Cell Signaling Technology, 50907S), anti-TBK1 (Cell Signaling Technology, 3504S), anti-Phospho-TBK1 (Ser172) (Cell Signaling Technology, 5483S), anti-IRF3 (Cell Signaling Technology, 11904S), anti-Phospho-IRF3 (Ser396) (Cell Signaling Technology, 4947S), anti-NF-κB p65 (Cell Signaling Technology, 8242S), anti-Phospho-NF-κB p65 (Ser536) (Cell Signaling Technology, 3033S), anti-TRIM22 (Invitrogen, PA5-51964), anti-SARS-CoV-2 nucleocapsid (GeneTex, GTX135357), and anti-tubulin (Sigma, T6199). Anti-SARS-CoV-2 Spike antibody (CR3022) was from Absolute Antibody. HRP-conjugated secondary antibodies (anti-mouse or anti-rabbit) were purchased from Amersham. Alexa Fluor-conjugated secondary antibodies were purchased from Invitrogen.
RNA isolation and RT-qPCR. Calu-3 cells (7×105 cells/well) or NHBE submersion cells (1×106 cells/well) were seeded into 6-well plates. The next day, if indicated, Calu-3 cells were pretreated with compounds for 1 hour at 37°C. Cells were inoculated with SARS-CoV-2 at the indicated MOI for the indicated time. Total RNA was isolated using TRIzol Reagent (Invitrogen) and purified using RNA Clean & Concentrator Kits (Zymo Research). cDNA was synthesized using 1 μg of total RNA, M-MLV Reverse Transcriptase (Invitrogen) and Random Primers (Invitrogen). Gene specific primers and Power SYBR Green PCR Master Mix (Applied Biosystems) were used to amplify cellular RNA and viral RNA by QuantStudio 6 Flex Real-Time PCR Systems (Applied Biosystems). The relative expression levels of target genes were calculated using the standard curve method and normalized to 18S ribosomal RNA as an internal control. Primers used in this study are listed in Table 1.
Immunoblotting. Calu-3 cells (7×105 cells/well) plated into 6-well plates were inoculated with SARS-CoV-2 (MOI of 0.5) or treated with diABZI or IFNs. At the indicated time points, cells were washed with PBS and lysed in RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% Sodium Deoxycholate, 0.1% SDS, 1% NP-40) supplemented with protease inhibitor cocktail (Sigma). Upon centrifugation at 13,000 rpm for 10 min at 4°C, clarified cell lysates were incubated with 6x sample buffer at 95°C for 10 min. Proteins were separated on 12% SDS-PAGE gels and transferred to Immobilon-P PVDF Transfer Membranes (Millipore). The membranes were blocked with 5% milk in TBS with 0.1% Tween 20 (TBST) for 1 hour at room temperature and incubated with primary antibodies overnight at 4°C. After 3 rinses with TBST, the membranes were incubated with Horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 hour at room temperature. Membranes were then developed with ECL Western Blotting Reagents (Amersham). Western blotting signals were visualized using Amersham Imager 680 (Amersham).
Confocal microscopy. Cells (1.5×105 cells/well) were seeded on collagen I coated glass coverslips (Electron Microscopy Sciences) into 24-well plates. After 24 hours, cells were inoculated with SARS-CoV-2 (MOI of 0.5) for 48 hours or treated with 10 μM diABZI for 30 min. Cells were fixed with 4% formaldehyde for 10 min and then blocked with 2% BSA in PBS with 0.1% Triton X-100 (PBST) for 1 hour at room temperature. Cells were incubated with the indicated primary antibodies overnight at 4°C. Coverslips were washed 3 times with PBST and then incubated with Hoechst 33342 (Sigma) and Alexa Fluor-conjugated secondary antibodies for 1 hour at room temperature. Following 3 rinses with PBST, coverslips were mounted on glass slides using VECTASHIELD Antifade Mounting Medium (Vector Laboratories). Cells were visualized using a 63x objective lens with a Leica DM5500Q confocal microscope.
NHBE ALI infections. NHBE-ALI cultures on inserts were visually inspected for integrity and cilia beating. Prior to infection, mucus was removed by gently rinsing the apical side with PBS. Drugs were added to the basolateral chamber 1 hour before virus inoculation on the apical surface with 100 μL of SARS-CoV-2 (MOI of 0.2) in PBS. One hour post infection, viral inoculum was removed, and the apical surface was rinsed with PBS. At 72 hpi, the inserts were processed for total RNA using Trizol, or fixed in 4% formaldehyde as described above. For microscopy, cells were stained with primary antibodies for >16 hours at 4°C and secondary antibodies for 2 hours at room temperature. Primary antibodies used are as follows: anti-SARS-CoV2 spike protein (rabbit, 1:100, GTX135356, GeneTex), anti-SARS-CoV2 nucleocapsid protein (rabbit, 1:100, GTX635679, GeneTex), anti-alpha tubulin (rat, 1:200, MA1-80017, ThermoFisher), anti-MUC5AC (mouse, 1:100, Clone 45M1, M5293, Sigma). Secondary antibodies from ThermoFisher were all diluted to 1:300 and are as follows: goat anti-mouse IgG (H+L) highly cross-adsorbed secondary antibody Alexa Fluor 488, goat anti-rabbit IgG (H+L) highly cross-adsorbed secondary antibody Alexa Fluor 568, goat anti-rat IgG (H+L) highly cross-adsorbed secondary antibody Alexa Fluor 647. Hoechst 33342 (10 μg/ml, ThermoFisher) was used to stain nuclei. After staining, membranes were removed from the insert using a surgical scalpel, and freed membranes subsequently mounted onto a glass coverslip and glass-bottom multi-well plates using ProLong Diamond Antifade Mountant (ThermoFisher). Mounted stained membranes were imaged on an automated confocal microscope system using a 10x objective (Opera Phenix HCS, Perkin Elmer). Full Z-stack image files were utilized for analysis of cell markers using Perkin Elmer’s Columbus Image Data Storage and Analysis System Columbus software. Sample tissue infection was calculated as a percentage of total cells present and normalized to DMSO control wells.
RNAseq. RNAseq Libraries for next-generation sequencing were prepared using the Illumina TruSeq stranded mRNA kit with Illumina TruSeq unique dual indices. Quality and quantity control of RNA and libraries were performed using an Agilent 4200 TapeStation and Qubit 4, respectively. Libraries were diluted to a final concentration of 4 nM and sequenced on an Illumina NextSeq 500 instrument to produce 75–base pair single end reads.
RNAseq analysis. Raw fastq files were trimmed to remove adapters and low quality reads with bbduk v38.79 (https://sourceforge.net/projects/bbmap/) using the parameters “ref=/bbmap/resources/adapters.fa ktrim=r k=23 mink=11 hdist=1 minlength=35 tpe tbo qtrim=r trimq=10”. Next transcripts were counted using salmon v1.2.1 (45) in pseudoalignment mode with the parameters “-l A–validateMappings–rangeFactorizationBins 4–seqBias”. Reads from diABZI-treated cells were aligned to the Homo sapiens GRCh38 (hg38) transcriptome. To avoid erroneous alignment of viral reads to host transcripts, reads from SARS-CoV-2 infected cells were mapped to a chimeric transcriptome comprised of the Homo sapiens GRCh38 (hg38) transcriptome and the SARS-CoV-2 transcriptome. The SARS-CoV-2 transcriptome was generated using the complete genome of the Wuhan-Hu-1 isolate (NC_045512.2) and high-confidence transcript annotations from Kim, et al. 2020 (46). Reads mapping to the SARS-CoV-2 transcriptome were then excluded for differential abundance analysis of host genes. Reads from mouse lung were mapped to the GRCm38.p6 mouse transcriptome. Transcriptomes and mouse-human homology tables were downloaded from Ensembl (47). Transcript counts were collapsed to the gene level using the R package tximport v1.16.1 (48) and differential abundance was analyzed using DESeq2 v1.28.1 (49). Heatmaps were generated with the ComplexHeatmap v2.4.3 (50) R package implementing Ward’s method (51) for clustering. Enrichment analysis was performed using the ClusterProfiler v3.16.1 (52) for GO term enrichment and ReactomePA v1.32.0 (53) for enrichment of Reactome pathway. P values were adjusted for multiple comparison using the FDR method. Data management was performed using base R (https://www.r-project.org/) and dplyr v1.0.2 (https://cran.r-project.org/package=dplyr).
Mouse experiments. Animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine (assurance number A3381–01). Virus inoculations were performed under anesthesia that was induced and maintained with ketamine hydrochloride and xylazine, and all efforts were made to minimize animal suffering. Heterozygous K18-hACE2 C57BL/6J mice (strain: B6.Cg-Tg(K18-ACE2)2Prlmn/J) were obtained from the Jackson Laboratory and were housed in groups and fed standard chow diets. Eight- to nine-week-old mice of both sexes were treated via an intranasal route with the indicated dose of diABZI or vehicle control at the indicated time point. For infections with WA1, the animals were infected with 103 PFU of SARS-CoV-2 by intranasal administration. For histology, lungs were harvested at 7 dpi and sections were stained with hematoxylin and eosin and imaged at 25x (left; scale bar, 2.5 mm), 100x (middle; scale bar, 500 μm), and 400x (right; scale bar, 100 μm) magnifications. Each image is representative of a group of 2-3 mice. For mouse studies with the South African strain, the spike gene of B.1.351 was cloned into the WA1/2020 infectious clone (30) to produce Wash-B.1.351 chimeric virus, as described previously (31). Eight-week-old female 129 mice (strain: 129S2/SvPasCrl) were obtained from Charles River Laboratories and were treated via the intranasal route with vehicle or 3 μg diABZI at the indicated time point. 1×105 FFU of the SARS-CoV-2 Wash-SA B.1.351 strain was administered by intranasal inoculation. All mice were sacrificed at 4 days post infection.
Measurement of viral burden. Tissues were weighed and homogenized with zirconia beads in a MagNA Lyser instrument (Roche Life Science) in 1 mL of DMEM media supplemented with 2% heat-inactivated FBS. Tissue homogenates were clarified by centrifugation at 10,000 rpm for 5 min and stored at −80°C. RNA was extracted using the MagMax mirVana Total RNA isolation kit (Thermo Scientific) on the Kingfisher Flex extraction robot (Thermo Scientific). RNA was reverse transcribed and amplified using the TaqMan RNA-to-CT 1-Step Kit (ThermoFisher). Reverse transcription was carried out at 48°C for 15 min followed by 2 min at 95°C. Amplification was accomplished over 40 cycles as follows: 95°C for 15 s and 60°C for 1 min. Copies of SARS-CoV-2 N gene RNA in samples were determined using a previously published assay (54). Briefly, a TaqMan assay was designed to target a highly conserved region of the N gene (Forward primer: ATGCTGCAATCGTGCTACAA; Reverse primer: GACTGCCGCCTCTGCTC; Probe: /56-FAM/TCAAGGAAC/ZEN/AACATTGCCAA/3IABkFQ/). This region was included in an RNA standard to allow for copy number determination down to 10 copies per reaction. The reaction mixture contained final concentrations of primers and probe of 500 and 100 nM, respectively.
diABZI RNAseq studies. C57BL/6 mice were inoculated by the intranasal route with 10 μg of diABZI in 40 μl PBS with 5% DMSO. Vehicle-treated mice received 40 μl of 5% DMSO in PBS. 6 and 12 hours post treatment, mice were sacrificed, and lungs were harvested. Lung tissues were snap-frozen in liquid nitrogen and transferred into 2 mL Lysing Matrix Tubes (MP Biomedicals) containing beads, followed by addition of 1 mL of TRIzol Reagent (Invitrogen). The samples were then homogenized using FastPrep-24 Classic bead beating grinder and lysis system (MP Biomedicals). 900 μL of TRIzol Reagent was added to 100 μL of homogenized tissues for RNA extraction and subsequent RNAseq analysis.
Cytokine and chemokine protein measurements. Lung homogenates were incubated with Triton-X-100 (1% final concentration) for 1 hour at room temperature to inactivate SARS-CoV-2. Homogenates then were analyzed for cytokines and chemokines by Eve Technologies Corporation (Calgary, AB, Canada) using their Mouse Cytokine Array/Chemokine Array 44-Plex (MD44) platform or Mouse Cytokine Array/Chemokine Array 31-Plex (MD31) platform.
Supplementary Materials
immunology.sciencemag.org/cgi/content/full/6/59/eabi9007/DC1
Fig. S1. SARS-CoV-2 evades early immune recognition.
Fig. S2. High-throughput screening identifies STING agonists as antivirals against SARS-CoV-2.
Fig. S3. STING agonist stimulates immune response.
Fig. S4. STING agonist restricts SARS-CoV-2 in NHBE-ALI cells.
Fig. S5. STING agonist induces ISGs in the mouse lung.
Fig. S6. Similarity of transcriptional responses in mouse lung and Calu-3 cells treated with diABZI.
Table S1. High throughput PAMP screen.
Data file S1. Raw data file (Excel spreadsheet)
Notes
Note added in proof: Another complementary study by Humphries et al. has shown that related STING agonists protect against SARS-CoV-2 infection (55).
- Copyright © 2021, American Association for the Advancement of Science