
Executing tumor cell death
There is increasing interest in developing cancer immunotherapies that target the innate immune pathways regulating cytokine production and cell death, but the interplay between these two closely connected processes is not well understood. In mouse and human cancer cell lines, Boehmer et al. demonstrate that cytokine production and apoptosis induced by retinoic acid–inducible gene I (RIG-I) ligands, including 5′-triphosphate RNA (3p-RNA), are two separable events in which RIG-I is required for production of type I interferon but not execution of apoptosis. Mass spectrometry and loss-of-function assays showed that 3p-RNA directly activates OAS1 and RNase L, which promoted translational arrest and depletion of antiapoptotic MCL-1. These results demonstrate that RIG-I–mediated apoptosis involves priming and effector stages, reminiscent of inflammasome activation, both of which could serve as potential targets for cancer immunotherapy.
Abstract
Cytoplasmic double-stranded RNA is sensed by RIG-I–like receptors (RLRs), leading to induction of type I interferons (IFN-Is), proinflammatory cytokines, and apoptosis. Here, we elucidate signaling mechanisms that lead to cytokine secretion and cell death induction upon stimulation with the bona fide RIG-I ligand 5′-triphosphate RNA (3p-RNA) in tumor cells. We show that both outcomes are mediated by dsRNA-receptor families with RLR being essential for cytokine production and IFN-I–mediated priming of effector pathways but not for apoptosis. Affinity purification followed by mass spectrometry and subsequent functional analysis revealed that 3p-RNA bound and activated oligoadenylate synthetase 1 and RNase L. RNase L–deficient cells were profoundly impaired in their ability to undergo apoptosis. Mechanistically, the concerted action of translational arrest triggered by RNase L and up-regulation of NOXA was needed to deplete the antiapoptotic MCL-1 to cause intrinsic apoptosis. Thus, 3p-RNA–induced apoptosis is a two-step process consisting of RIG-I–dependent priming and an RNase L–dependent effector phase.
INTRODUCTION
The innate immune system is the first line of defense against invading pathogens. Activation of the innate immune system in infected cells mainly results in two events: (i) induction of antiviral cytokines to alarm neighboring cells and to attract and activate immune cells and (ii) activation of cell-intrinsic defense mechanisms, which may ultimately lead to programmed cell death to limit replication of intracellular pathogens. The pathways of cytokine induction and cell death are highly interconnected and tightly regulated on a molecular level (1). Because erroneous induction of programmed cell death can have fatal consequences (2, 3), multicellular organisms have evolved “safety switches” to prevent inappropriate cell death from being executed. One common principle most prominently described in the context of inflammasomes (4) is a two-step mechanism consisted of a “priming” and an “effector” step by which these pathways are activated.
Viral RNAs can bind to and activate innate immune receptors such as the retinoic acid–inducible gene I (RIG-I)–like receptors (RLRs). Short double-stranded RNAs (dsRNAs) with a 5′-triphosphate moiety [5′-triphosphate RNA (3p-RNA)] are well established as specific ligands for RIG-I (5–7). Extensive research has characterized the molecular signaling events underlying type I interferon (IFN-I) and cytokine production: Upon binding of 3p-RNA, RIG-I activates downstream IFN regulatory factor 3 (IRF3), mitogen-activated protein kinase, and nuclear factor κB pathways via mitochondrial antiviral-signaling protein (MAVS) (8). Besides its essential role in antiviral cytokine induction, RIG-I has been shown by several studies to activate programmed cell death in response to viral RNA, with some selectivity for tumor cells. Initial reports describing melanoma cell–selective apoptosis found IFN-I–independent induction of the proapoptotic Bcl-2 homology (BH)3-only proteins p53-up-regulated modulator of apoptosis (PUMA) and phorbol-12-myristate-13-acetate-induced protein 1 (NOXA) to cause intrinsic apoptosis (9–11). Other reports suggested several other mechanistic models: TNF-related apoptosis-inducing ligand (TRAIL) signaling (12), down-regulation of Bcl-2/baculoviral IAP repeat-containing protein 3 (BIRC3)/protein kinase C epsilon type (PRKCE) (13), activation of caspase-8 via MAVS (14), and RLR-induced IRF3-mediated pathway of apoptosis (15, 16). However, even central aspects of the signaling involved in RIG-I–dependent cell death induction such as the requirement for IFN-I signaling are elusive.
We and others (9–11, 17–20) showed that 3p-RNA has strong antitumoral activity through induction of IFN-I and cancer cell–selective apoptosis. Recently, we found this cell death to be highly immunogenic, leading to potent adaptive antitumoral immune responses (17, 21). A phase 1 clinical trial of a RIG-I agonist for the treatment of solid tumors is ongoing (NCT03739138), highlighting the need to improve our understanding of 3p-RNA–induced cell death. Specifically in the context of cancers, well-defined genetic evidence discriminating signaling mechanisms that cause apoptosis from those that cause cytokine induction in response to 3p-RNA exposure is still lacking. Here, we provide evidence that 3p-RNA elicits cell death via a two-step mechanism involving RIG-I/IFN-I–mediated priming of 3p-RNA–sensing executioner pathways and proapoptotic mitochondrial priming of apoptosis. Combining unbiased RNA sequencing (RNA-seq) and proteomic approaches, we characterize the 3p-RNA sensor proteins and analyze in depth the molecular mechanisms of the priming and effector phases.
RESULTS
RIG-I ligands execute tumor cell death by a RIG-I–independent, IFN-I–primed mechanism
To distinguish pathways inducing cytokine production, which might affect cell death in an autocrine or paracrine manner, from cell-intrinsic proapoptotic mechanisms, we first generated human 1205Lu and mouse B16 melanoma cell lines deficient for RIG-I (encoded by DDX58 in the human and Ddx58 in the mouse), melanoma differentiation-associated protein 5 (MDA5) (only in 1205Lu cells, encoded by IFIH1 in the human), and MAVS (encoded by MAVS in the human and Mavs in the mouse) using CRISPR-Cas9–mediated gene editing. In line with previous findings (10), we observed that 3p-RNA strongly induced cell death, as measured by annexin A5 assay, and IFN-I signaling, as measured by IFN-I–dependent major histocompatibility complex class I (MHC-I) surface expression and IFN-β expression, compared with a nonstimulatory control RNA (CTRL) in wild-type (WT) cells (Fig. 1, A and B, and fig. S1, A to C). In addition, cells deficient of RIG-I or its adaptor protein MAVS did not show any induction of cell death or cytokines, whereas MDA5 was dispensable for 3p-RNA–mediated signaling, confirming 3p-RNA to be a highly specific ligand for RIG-I in both human and murine melanoma cells. To assess how secreted factors may influence cell death induction, we next cocultured WT and RIG-I–deficient cells. Fluorescent labeling of either of the cell lines before seeding allowed their discrimination by flow cytometry (Fig. 1C). Notably, the ability to undergo apoptosis in response to 3p-RNA treatment was restored in RIG-I–deficient 1205Lu and B16 cells under coculture conditions (Fig. 1D and fig. S1D). This finding demonstrates that cell death and cytokine induction are two separable events upon the detection of 3p-RNA. Furthermore, this suggests the existence of one or more factors secreted by WT cells upon RIG-I activation that restore the ability of 3p-RNA to induce cell death in RIG-I–deficient cells.
(A) 1205Lu human melanoma cells of the indicated genotype were stimulated with 80 nM 3p-RNA or nonstimulatory control RNA (CTRL) and analyzed for cell death by annexin A5 assay and for MHC-I expression after 48 hours (n = 2). MFI, median fluorescence intensity. (B) ELISA for human IFN-β in the cell supernatant after stimulation with 160 nM 3p-RNA or controls for 48 hours (n = 3). (C) Schematic of coculture assay and gating strategy for flow cytometric analysis. (D) WT and RIG-I–deficient 1205Lu cells were cultured alone or cocultured as depicted in (C), treated with 160 nM 3p-RNA or CTRL, and analyzed by flow cytometry (n = 3, Student’s t test). (E) 1205Lu cells of indicated genotype were prestimulated with IFN-α (1000 U/ml) 18 hours before transfection with 40, 80, or 160 nM 3p-RNA or CTRL. Viability was assessed by CTB assay 48 hours after RNA transfection (n = 3, Student’s t test). (F) WT and RIG-I–deficient or WT and RIG-I/IFNAR1–deficient 1205Lu cells were cultured alone or cocultured, treated with 160 nM 3p-RNA or CTRL, and analyzed by flow cytometry (n = 3, one-way analysis of variance (ANOVA) with Sidak’s multiple comparisons test; asterisks indicate results of post hoc test.) (G) WT and IRF3−/− 1205Lu cells were prestimulated with IFN-α (1000 U/ml) or mock for 18 hours before transfection of indicated RNAs as shown (160 nM). Viability was measured by CTB assay (n = 3, one-way ANOVA with Dunnett’s multiple comparisons test). (H) Viability of WT and IFNAR1−/− 1205Lu cells 48 hours after transfection with 3p-RNA or CTRL (160 nM) (n = 3, Student’s t test). (I) CTB assay of WT and DDX58−/− cells transfected with total RNA isolated from WT or M51R VSV-infected cells (1 μg/ml) for 48 hours. DDX58−/− cells were treated with IFN-α or mock (ø) for 18 hours before RNA transfection [n = 2, one-way ANOVA (WT) or two-way ANOVA (DDX58−/−), followed by Sidak’s multiple comparisons test]. Asterisks indicate results of post hoc test. Asterisks on bars represent comparison to CTRL-RNA. Means + SEM of independent experiments are shown; *P < 0.05, **P < 0.01, and ***P < 0.001; ns, not significant.
” data-icon-position=”” data-hide-link-title=”0″>
(A) 1205Lu human melanoma cells of the indicated genotype were stimulated with 80 nM 3p-RNA or nonstimulatory control RNA (CTRL) and analyzed for cell death by annexin A5 assay and for MHC-I expression after 48 hours (n = 2). MFI, median fluorescence intensity. (B) ELISA for human IFN-β in the cell supernatant after stimulation with 160 nM 3p-RNA or controls for 48 hours (n = 3). (C) Schematic of coculture assay and gating strategy for flow cytometric analysis. (D) WT and RIG-I–deficient 1205Lu cells were cultured alone or cocultured as depicted in (C), treated with 160 nM 3p-RNA or CTRL, and analyzed by flow cytometry (n = 3, Student’s t test). (E) 1205Lu cells of indicated genotype were prestimulated with IFN-α (1000 U/ml) 18 hours before transfection with 40, 80, or 160 nM 3p-RNA or CTRL. Viability was assessed by CTB assay 48 hours after RNA transfection (n = 3, Student’s t test). (F) WT and RIG-I–deficient or WT and RIG-I/IFNAR1–deficient 1205Lu cells were cultured alone or cocultured, treated with 160 nM 3p-RNA or CTRL, and analyzed by flow cytometry (n = 3, one-way analysis of variance (ANOVA) with Sidak’s multiple comparisons test; asterisks indicate results of post hoc test.) (G) WT and IRF3−/− 1205Lu cells were prestimulated with IFN-α (1000 U/ml) or mock for 18 hours before transfection of indicated RNAs as shown (160 nM). Viability was measured by CTB assay (n = 3, one-way ANOVA with Dunnett’s multiple comparisons test). (H) Viability of WT and IFNAR1−/− 1205Lu cells 48 hours after transfection with 3p-RNA or CTRL (160 nM) (n = 3, Student’s t test). (I) CTB assay of WT and DDX58−/− cells transfected with total RNA isolated from WT or M51R VSV-infected cells (1 μg/ml) for 48 hours. DDX58−/− cells were treated with IFN-α or mock (ø) for 18 hours before RNA transfection [n = 2, one-way ANOVA (WT) or two-way ANOVA (DDX58−/−), followed by Sidak’s multiple comparisons test]. Asterisks indicate results of post hoc test. Asterisks on bars represent comparison to CTRL-RNA. Means + SEM of independent experiments are shown; *P < 0.05, **P < 0.01, and ***P < 0.001; ns, not significant.
IFN-I are cytokines released upon 3p-RNA stimulation that potently induce an antiviral state by regulating hundreds of genes (22, 23), making them likely candidates for priming cells for RNA-induced death. To address this hypothesis, we stimulated DDX58−/− and MAVS−/− cells with recombinant IFN-α2a (IFN-α) before 3p-RNA transfection. 3p-RNA induced death in the two RLR signaling–deficient human melanoma cell lines 1205Lu (Fig. 1E) and C8161 (fig. S1E) prestimulated with IFN-α. IFN-α stimulation alone or together with CTRL transfection did not lead to cell death, ruling out a direct cytotoxic effect of IFN-α or RNA transfection. To determine whether IFN-I was essential for priming RIG-I–deficient cells in the coculture setup, we used cells deficient in RIG-I and the IFN-α/β receptor 1 (IFNAR1). In contrast to DDX58−/− cells, DDX58−/−IFNAR1−/− cells did not undergo cell death upon coculture with WT cells (Fig. 1F). The absence of MHC-I up-regulation confirmed that IFN-I signaling was disrupted (fig. S1F). To further examine the role of IFN-I signaling, we generated knockout (KO) cells for IRF3, encoding the main transcription factor that drives the induction of IFN-I by RLRs (22, 24). Besides its role in IFN-I induction, IRF3 has been implicated in dsRNA-induced apoptosis by directly interacting with Bcl-2-associated X protein (BAX) (25). Cell death induction by 3p-RNA was strongly reduced in IRF3−/− cells but could be rescued by the addition of exogenous IFN-α (Fig. 1G and fig. S1G). These findings argue against a direct involvement of IRF3 in 3p-RNA–induced apoptosis but suggest IRF3 to be needed for transcriptional priming through IFN-I. In line with these findings, IFNAR1-deficient cells or cells incubated with an IFNAR1-blocking antibody displayed markedly decreased 3p-RNA–induced cell death, despite intact RLR and IRF3 signaling (Fig. 1H and fig. S1H).
To exclude these findings being specific to the synthetic 3p-RNA, we extended our investigation to ligands of RIG-I that are produced during viral infection. We isolated RNA from vesicular stomatitis virus (VSV)–infected cells, transfected it into WT and RIG-I–deficient cells, and measured cellular viability after 48 hours (Fig. 1I). VSV-derived RNAs significantly reduced cell viability compared with control RNA isolated from uninfected cells. Again, RIG-I–deficient cells only showed pronounced induction of cell death when stimulated with IFN-α before RNA transfection. Analysis of C-X-C motif chemokine 10 (CXCL10) secretion from WT cells confirmed that VSV-derived RNAs were immunostimulatory (fig. S1I).
These findings indicate that IFN-I signaling is critical for an effective induction of cell death by 3p-RNA and natural RIG-I ligands. Furthermore and in contrast to early publications, RIG-I signaling itself seems to be dispensable for cell death when exogenous IFN-I is provided, suggesting the existence of a distinct IFN-I–inducible 3p-RNA–sensing pathway that executes cell death.
3p-RNA specifically binds oligoadenylate synthetase 1 and activates latent endoribonuclease
We next asked which dsRNA-sensing pathway executes cell death in response to 3p-RNA. To gain further insight into the events downstream of 3p-RNA stimulation in WT and RIG-I–deficient 1205Lu cells, we analyzed the transcriptome after stimulation with 3p-RNA and/or IFN-α by RNA-seq. Six hours after transfection with 3p-RNA, WT cells showed significant changes of mRNA levels of more than 4000 genes compared with an unstimulated control (Fig. 2A). In contrast, not a single gene was differentially expressed by 3p-RNA treatment alone in DDX58−/− cells. However, IFN-α priming for 18 hours and subsequent 3p-RNA treatment of DDX58−/− cells significantly changed the expression of more than 6000 genes compared with cells treated with IFN-α alone. Analysis of Gene Ontology molecular function (GO:MF) terms of those genes revealed an enrichment of genes in transcriptional regulation (fig. S2A). However, because of the lack of known gene signatures associated with RNA recognition, we were not able to infer upstream pathways. Therefore, we analyzed RNA-binding protein genes that were selectively up-regulated in response to IFN-α by performing RNA-seq of 1205Lu WT cells untreated or treated with IFN-α for 4 hours. IFN-α treatment led to a significant up-regulation of 788 genes, among which 71 were annotated with the GO:MF terms “double-stranded RNA binding” or “RNA binding” (fig. S2B). Among the 30 genes with the highest log2 fold changes (FCs) (Fig. 2B), we found a group of RNA sensors known to mediate antiviral defense by directly acting on foreign nucleic acids such as protein kinase R (PKR), oligoadenylate synthetase 1 (OAS1) to OAS3, and the IFN-induced protein with tetratricopeptide repeats (IFIT) family (26). The 5′-triphosphate moiety of viral RNA species is a potent pathogen-associated molecular pattern for their detection by the host and their discrimination from endogenous RNA (5, 27, 28). We further validated the necessity of the 5′-triphosphate moiety for RIG-I–independent cell death by stimulating IFN-α–primed 1205Lu DDX58−/− cells with dephosphorylated 3p-RNA [OH-RNA (FastAP)] and measured cytotoxicity (Fig. 2C and fig. S2C). Cell death induction by 3p-RNA was markedly reduced after FastAP treatment. Besides RIG-I itself, only IFIT1 and, to some extent, PKR (encoded by EIF2AK2) have been described to preferentially bind RNA with a 5′-triphosphate moiety (28–30). However, studying KO cell lines demonstrated that both IFIT1 and PKR are dispensable for cell death induction by 3p-RNA (fig. S3, A and B). IFIT1−/− cells showed a more rapid cell death induction compared with WT and other KO cells (fig. S3B).
(A) Volcano plots of differential gene expression (log2 FC) for indicated conditions upon 3p-RNA treatment. (B) Top 30 induced genes (log2 FC) annotated with the GO:MF term double-stranded RNA binding (blue) or RNA binding (gray) as determined by RNA-seq in 1205Lu DDX58−/− cells stimulated for 4 hours with IFN-α compared with untreated control. (C) IFN-α–primed 1205Lu DDX58−/− cells were transfected with FastAP- or mock-treated 3p-RNA as indicated. Cell death induction was measured 48 hours after transfection (n = 3, Student’s t test). (D) Volcano plot of relative LFQ of proteins bound to 3p-RNA-biot compared with FastAP-treated 3p-RNA-biot (OH-RNA-biot) as quantified by LC-MS/MS (n = 5). Proteins up-regulated in (A) with GO:MF term double-stranded RNA binding are highlighted in blue, those significantly enriched are indicated in green. (E) Western blot validation of MS results. 3p-RNA-biot or OH-RNA-biot was bound to streptavidin beads and used for affinity purification (AP) of proteins in lysates from unstimulated or IFN-α–stimulated (18 hours) cells. Thirty micrograms of protein lysate was used as input. Representative of three independent experiments. (F) OAS1 activation by chemically synthesized RNAs as measured by pyrophosphate production using a chromogenic assay. CTRL, non-immunostimulatory control RNA; 3p-RNA/OH-RNA, custom-synthesized RNAs with (3p) or without (OH) 5′-triphosphate group; 3′-ssPy, dsRNA with single-stranded 3′-pyrimidine overhang (mean ± SD of n = 3, one-way ANOVA, followed by Dunnett’s multiple comparisons test; asterisks indicate results of post hoc test). (G and H) rRNA integrity analysis of total RNA isolated from 1205Lu DDX58−/− cells using RNA BioAnalyzer. (G) IFN-α–primed cells were transfected with OH-RNA, 3p-RNA (both 80 nM) or poly(I:C) (1 μg/ml) for 6 hours. Left: Representative image of four independent experiments. Right: Quantification of RNA degradation by RNA-integrity number (RIN) (n = 4, Student’s t test). (H) Cells untreated or treated with IFN-α (18 hours), 3p-RNA (6 hours), or IFN-α and then 3p-RNA. Representative of at least three independent experiments. Unless stated otherwise, means + SEM of independent experiments are shown; *P < 0.05, **P < 0.01, and ****P < 0.0001.
” data-icon-position=”” data-hide-link-title=”0″>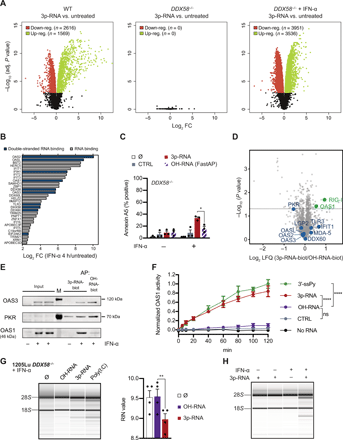
(A) Volcano plots of differential gene expression (log2 FC) for indicated conditions upon 3p-RNA treatment. (B) Top 30 induced genes (log2 FC) annotated with the GO:MF term double-stranded RNA binding (blue) or RNA binding (gray) as determined by RNA-seq in 1205Lu DDX58−/− cells stimulated for 4 hours with IFN-α compared with untreated control. (C) IFN-α–primed 1205Lu DDX58−/− cells were transfected with FastAP- or mock-treated 3p-RNA as indicated. Cell death induction was measured 48 hours after transfection (n = 3, Student’s t test). (D) Volcano plot of relative LFQ of proteins bound to 3p-RNA-biot compared with FastAP-treated 3p-RNA-biot (OH-RNA-biot) as quantified by LC-MS/MS (n = 5). Proteins up-regulated in (A) with GO:MF term double-stranded RNA binding are highlighted in blue, those significantly enriched are indicated in green. (E) Western blot validation of MS results. 3p-RNA-biot or OH-RNA-biot was bound to streptavidin beads and used for affinity purification (AP) of proteins in lysates from unstimulated or IFN-α–stimulated (18 hours) cells. Thirty micrograms of protein lysate was used as input. Representative of three independent experiments. (F) OAS1 activation by chemically synthesized RNAs as measured by pyrophosphate production using a chromogenic assay. CTRL, non-immunostimulatory control RNA; 3p-RNA/OH-RNA, custom-synthesized RNAs with (3p) or without (OH) 5′-triphosphate group; 3′-ssPy, dsRNA with single-stranded 3′-pyrimidine overhang (mean ± SD of n = 3, one-way ANOVA, followed by Dunnett’s multiple comparisons test; asterisks indicate results of post hoc test). (G and H) rRNA integrity analysis of total RNA isolated from 1205Lu DDX58−/− cells using RNA BioAnalyzer. (G) IFN-α–primed cells were transfected with OH-RNA, 3p-RNA (both 80 nM) or poly(I:C) (1 μg/ml) for 6 hours. Left: Representative image of four independent experiments. Right: Quantification of RNA degradation by RNA-integrity number (RIN) (n = 4, Student’s t test). (H) Cells untreated or treated with IFN-α (18 hours), 3p-RNA (6 hours), or IFN-α and then 3p-RNA. Representative of at least three independent experiments. Unless stated otherwise, means + SEM of independent experiments are shown; *P < 0.05, **P < 0.01, and ****P < 0.0001.
We next used an unbiased proteomic approach by performing affinity purification of proteins from IFN prestimulated 1205Lu lysates using biotinylated 3p-RNA (3p-RNA-biot) or a dephosphorylated control (OH-RNA-biot), followed by mass spectrometry (MS) and label-free quantification (LFQ). Among the top 30 up-regulated proteins with RNA binding function, besides RIG-I that served as the internal positive control, only OAS1 was significantly enriched by 3p-RNA-biot compared with OH-RNA-biot (n = 5) (Fig. 2D). Western blot analysis confirmed the higher affinity of OAS1 to 3p-RNA-biot and revealed that IFN-I induction was required for the detection of OAS1 and OAS3 and for their binding to RNA in these cells, whereas PKR already bound RNA in the steady-state condition and, in our study, bound 3p-RNA-biot less efficiently than OH-RNA-biot (Fig. 2E).
OAS1, OAS2, and OAS3 synthesize the second messenger 2′-5′ oligoadenylate (2-5A) in response to activation by viral dsRNA (31, 32). To validate that OAS1 is directly activated in a 5′-triphosphate–dependent manner, we measured pyrophosphate production by recombinant human OAS1 as a reaction by-product of 2-5A synthesis using a chromogenic assay as previously described (Fig. 2F) (33). Addition of 3p-RNA showed potent activation of OAS1, comparable to a dsRNA carrying a 3′–single-stranded pyrimidine (3′-ssPy) motif, a known potentiator of OAS1 activation (33), whereas the nonphosphorylated control RNA (OH-RNA) did not show significant levels of OAS1 activity.
2-5A mediates dimerization, and thereby activation, of the latent endoribonuclease (RNase L, encoded by RNASEL) (34, 35). The hallmark of OAS/RNase L activation is the degradation of viral and cellular RNA, including ribosomal RNA (rRNA) (36, 37). To test whether RNase L gets activated in response to 3p-RNA treatment, we studied rRNA integrity in RIG-I–deficient cells. 3p-RNA caused degradation of cellular RNA in IFN-α–primed cells, whereas OH-RNA did not (Fig. 2G). Furthermore, only 3p-RNA transfection in combination with IFN-α priming led to degradation of rRNA (Fig. 2H), indicating that RNase L can get activated only after IFN-I–dependent priming, likely involving recognition of 3p-RNA by OAS.
These results together exclude known 3p-RNA sensors to be responsible for cell death induction. Instead, we reveal a 3p-RNA–specific binding and activation characteristic of OAS1 and show the activation of RNase L after 3p-RNA transfection (Fig. 2, D to H).
The OAS1/RNase L pathway mediates cell death induced by 3p-RNA
To investigate whether the activation of OAS/RNase L is essential for cell death induced by 3p-RNA, we generated RNASEL−/− and DDX58−/−RNASEL−/− cells and assessed RNA degradation after 3p-RNA stimulation, with or without IFN-α pretreatment (Fig. 3A and fig. S4A). The RNA degradation in WT or DDX58−/− cells upon stimulation with 3p-RNA entirely depended on the presence of RNase L. In addition, RNASEL−/− cells were highly resistant to 3p-RNA–induced cell death, which was particularly pronounced at higher RNA concentrations (Fig. 3B and fig. S4B). Cell death assay showed complete resistance of IFN-α–primed DDX58−/−RNASEL−/− cells over the course of 48 hours of 3p-RNA treatment (Fig. 3C). RNase L has been described to strengthen the IFN-I response through sensing RNA cleavage products by RLRs (38). To rule out a defect in IFN-I–mediated priming in RNase L–deficient cells, we investigated a coculture setup of WT, Mavs−/−, Rnasel−/−, or Mavs−/−Rnasel−/− B16 cells, ensuring equal IFN-I signaling in cells with all genotypes. As expected, Mavs−/− cells did not show a significant difference compared with WT cells upon coculture, indicating effective priming (fig. S4C). In contrast, Rnasel−/− and Mavs−/−Rnasel−/− cells showed significantly reduced cell death.
(A) rRNA integrity analysis of total RNA isolated from 1205Lu cells with indicated genotypes untreated or treated with IFN-α (1000 U/ml, 18 hours), transfected with 3p-RNA (12 hours, 160 nM), or treated with IFN-α (18 hours), and then with 3p-RNA (12 hours). Representative of two independent experiments. (B) Cell death induction in 1205Lu WT and RNASEL−/− cells treated with increasing doses of 3p-RNA (40, 80, and 160 nM) for 48 hours (n = 4, Student’s t test). (C) Kinetic analysis of cell death induction using xCELLigence real-time cell analyzer. WT and indicated KOs in 1205Lu cells were plated on an E-Plate 96 PET with or without IFN-α (1000 U/ml) for 16 hours and then transfected with 3p-RNA (160 nM). Impedance was analyzed for 48 hours after 3p-RNA transfection. Mean of n = 3. (D) Western blot analysis of 1205Lu DDX58−/− cells overexpressing OAS1, OAS2, or OAS3 or empty vector control with or without 18-hour stimulation with Dox (1 μg/ml) or IFN-α (1000 U/ml). Representative of two independent experiments. Relative transgene expression to IFN-α–stimulated sample after normalization to loading control is shown below. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (E) CTB assay of cells as in (D) with or without stimulation with either IFN-α (1000 U/ml) or Dox (0.01, 0.1, and 1 μg/ml) and treatment with 160 nM 3p-RNA for 48 hours (n = 5, Student’s t test). (F) Viability of 1205Lu WT and OAS1−/− cells 48 hours after RNA transfection with 160 nM, normalized to untreated (n = 3, Student’s t test). (G) CTB assay of 1205Lu WT and RNASEL−/− cells treated with total RNA (1 μg/ml) from cells infected with indicated VSV or SeV for 48 hours [n = 7 (CTRL-RNA), 5 (VSV-M51R), and 4 (VSV-WT and SeV), Student’s t test]. (H) Flow cytometric analysis of viability of WT or RNase L–deficient B16F10 cells 48 hours after transfection (n = 3, Student’s t test). RNA preparations as described in (G) were used. Unless stated otherwise, means + SEM of independent experiments are shown; *P < 0.05, **P < 0.01, and ***P < 0.001.
” data-icon-position=”” data-hide-link-title=”0″>
(A) rRNA integrity analysis of total RNA isolated from 1205Lu cells with indicated genotypes untreated or treated with IFN-α (1000 U/ml, 18 hours), transfected with 3p-RNA (12 hours, 160 nM), or treated with IFN-α (18 hours), and then with 3p-RNA (12 hours). Representative of two independent experiments. (B) Cell death induction in 1205Lu WT and RNASEL−/− cells treated with increasing doses of 3p-RNA (40, 80, and 160 nM) for 48 hours (n = 4, Student’s t test). (C) Kinetic analysis of cell death induction using xCELLigence real-time cell analyzer. WT and indicated KOs in 1205Lu cells were plated on an E-Plate 96 PET with or without IFN-α (1000 U/ml) for 16 hours and then transfected with 3p-RNA (160 nM). Impedance was analyzed for 48 hours after 3p-RNA transfection. Mean of n = 3. (D) Western blot analysis of 1205Lu DDX58−/− cells overexpressing OAS1, OAS2, or OAS3 or empty vector control with or without 18-hour stimulation with Dox (1 μg/ml) or IFN-α (1000 U/ml). Representative of two independent experiments. Relative transgene expression to IFN-α–stimulated sample after normalization to loading control is shown below. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (E) CTB assay of cells as in (D) with or without stimulation with either IFN-α (1000 U/ml) or Dox (0.01, 0.1, and 1 μg/ml) and treatment with 160 nM 3p-RNA for 48 hours (n = 5, Student’s t test). (F) Viability of 1205Lu WT and OAS1−/− cells 48 hours after RNA transfection with 160 nM, normalized to untreated (n = 3, Student’s t test). (G) CTB assay of 1205Lu WT and RNASEL−/− cells treated with total RNA (1 μg/ml) from cells infected with indicated VSV or SeV for 48 hours [n = 7 (CTRL-RNA), 5 (VSV-M51R), and 4 (VSV-WT and SeV), Student’s t test]. (H) Flow cytometric analysis of viability of WT or RNase L–deficient B16F10 cells 48 hours after transfection (n = 3, Student’s t test). RNA preparations as described in (G) were used. Unless stated otherwise, means + SEM of independent experiments are shown; *P < 0.05, **P < 0.01, and ***P < 0.001.
We further investigated the importance of OAS-specific up-regulation compared with the induction of other IFN-I–stimulated genes and analyzed the individual contribution of OAS1, OAS2, and OAS3 in 3p-RNA sensing. We generated cell lines with doxycycline (Dox)–inducible overexpression of OAS1, OAS2, or OAS3 (Fig. 3D) to bypass the need for IFN-α prestimulation for the up-regulation of these proteins. Subsequent transfection with 3p-RNA or control RNA showed that overexpression of OAS1 or, to a lesser extent, OAS2 was sufficient to induce cell death upon 3p-RNA transfection (Fig. 3E). OAS1 induction in combination with 3p-RNA transfection led to a robust activation of RNase L as monitored by specific rRNA degradation patterns (fig. S4D). In addition to the gain-of-function experiments, we used OAS1-deficient 1205Lu cells to examine the importance of OAS1 in cells that harbor intact RIG-I signaling. OAS1 deficiency caused a strong reduction of cell death upon transfection with 3p-RNA (Fig. 3F).
To expand our results to natural ligands of RIG-I, we isolated total RNA from cells infected with different negative-strand RNA viruses and transfected them into WT or RNase L–deficient cells. RNase L deficiency significantly protected cells from death upon transfection of RNA isolated from cells infected with VSV-WT, VSV-M51R, or Sendai virus (SeV) (Fig. 3, G and H). These data indicate that natural RIG-I ligands similarly induce RNase L–dependent apoptosis.
Collectively, these data demonstrate that the OAS/RNase L system triggers cell death in response to synthetic and natural RIG-I ligands. Despite apparent redundancies within the OAS protein family, OAS1 seems to be the main sensor for 3p-RNA upstream of RNase L activation.
3p-RNA induces intrinsic apoptosis
To characterize the pathways leading to cell death in the presence or absence of RIG-I signaling, we analyzed the activation of cell death pathways that are reported to be engaged in response to viral dsRNA. We first measured the activation of caspases, one hallmark of apoptosis activation (39), in response to 3p-RNA treatment by a fluorescently labeled caspase probe. As expected, the activation of caspases was highly dependent on intact RIG-I signaling in the absence of exogenous IFN-α (Fig. 4A). Prestimulation of cells deficient in RIG-I or MAVS with IFN-α, however, rescued their ability to activate caspases. Using the pancaspase inhibitor z-VAD-fmk (z-VAD), we saw a strong dependence of cell death induction by 3p-RNA on caspases in WT and DDX58−/− cells (Fig. 4B).
(A) 1205Lu cells of indicated genotypes were transfected with 3p-RNA with or without prestimulation with IFN-α (1000 U/ml) for 18 hours. Caspase-positive, fixable viability dye (FVD)–negative cells were measured using Live Cell Caspase Probe via flow cytometry 30 hours after transfection (n = 3, Student’s t test). (B) 1205Lu cells were incubated with the pancaspase inhibitor z-VAD (20 μM, + or 80 μM, ++) or solvent control for 1 hour before RNA transfection, and cell death induction was measured after 48 hours (n = 2). (C) Western Blots of BAX and BAK expression in WT and RIG-I–deficient cells treated with sgRNAs targeting BAX and BAK1 (sgBAX/BAK) or an irrelevant control (sgCTRL). Representative of two independent experiments. (D) Cell death induction by 3p-RNA treatment (160 nM) 48 hours after transfection in IFN-α–primed DDX58−/− or WT 1205Lu cells (n = 2, Student’s t test). Means + SEM of independent experiments are shown; *P < 0.05, **P < 0.01, and ***P < 0.001.
” data-icon-position=”” data-hide-link-title=”0″>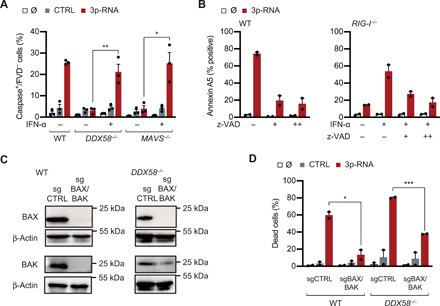
(A) 1205Lu cells of indicated genotypes were transfected with 3p-RNA with or without prestimulation with IFN-α (1000 U/ml) for 18 hours. Caspase-positive, fixable viability dye (FVD)–negative cells were measured using Live Cell Caspase Probe via flow cytometry 30 hours after transfection (n = 3, Student’s t test). (B) 1205Lu cells were incubated with the pancaspase inhibitor z-VAD (20 μM, + or 80 μM, ++) or solvent control for 1 hour before RNA transfection, and cell death induction was measured after 48 hours (n = 2). (C) Western Blots of BAX and BAK expression in WT and RIG-I–deficient cells treated with sgRNAs targeting BAX and BAK1 (sgBAX/BAK) or an irrelevant control (sgCTRL). Representative of two independent experiments. (D) Cell death induction by 3p-RNA treatment (160 nM) 48 hours after transfection in IFN-α–primed DDX58−/− or WT 1205Lu cells (n = 2, Student’s t test). Means + SEM of independent experiments are shown; *P < 0.05, **P < 0.01, and ***P < 0.001.
In the setting of RLR-induced apoptosis, both intrinsic and extrinsic pathways have been shown to be involved (10, 12, 15). Intrinsic apoptosis is induced by an imbalance of pro- and antiapoptotic B-cell lymphoma 2 (BCL2) family members toward proapoptotic, facilitating BAX/Bcl-2 homologous antagonist/killer (BAK)1 pore formation in the outer mitochondrial membrane and subsequent cytochrome c release to the cytosol. To test the involvement of BAX/BAK in apoptosis induction by 3p-RNA, we targeted genes encoding these proteins in WT and DDX58−/− 1205Lu cells using CRISPR-Cas9 and evaluated the efficiency of KO on protein level by Western blotting (Fig. 4C). Both WT and DDX58−/− cells showed profoundly impaired ability to undergo apoptosis upon 3p-RNA transfection after genetically targeting BAX/BAK (Fig. 4D), with the extent of cell death reduction correlating to the KO efficacy observed in Fig. 4C. Thus, intrinsic apoptosis is the main cell death pathway engaged by 3p-RNA treatment in WT and DDX58−/− cells in vitro.
RNase L licenses myeloid cell leukemia 1 depletion through NOXA up-regulation and differential mRNA degradation
We then investigated by what molecular mechanism RNase L executes cell death and how this mechanism can be related with the described requirement of proapoptotic NOXA expression. The main biological result of RNase L activation within cells is the rapid arrest of translation (40). Using an antipuromycin antibody, we monitored de novo protein synthesis by measuring puromycin incorporation into peptide chains after 3p-RNA treatment for the whole proteome (Fig. 5A) and cell surface proteins [termed surface sensing of translation (SUnSET) (41); fig. S5A]. WT cells showed a substantial reduction in translational activity after treatment with 3p-RNA, comparable to the complete block of translation mediated by cycloheximide (CHX) (Fig. 5A and fig. S5A). This effect was lost in RNase L–deficient cells, indicating RNase L–dependent translational arrest to be engaged upon 3p-RNA treatment.
(A) Measurement of puromycin (Puro) incorporation detected by anti–puromycin (Puro) blot (top image) and total protein (Stain-Free technology, bottom image) in IFN-α–primed 1205Lu WT or RNASEL−/− cells treated with CTRL, 3p-RNA (both 160 nM), or CHX (10 μg/ml) for 6 hours. Representative of two independent experiments. (B) Cell death induction in 1205Lu WT cells 24 hours after stimulation with small-molecule inhibitors of BCL2 family proteins (all used at 1 μM) in the presence or absence of CHX (1 μg/ml) [n = 2 to 4, two-way ANOVA, followed by Tukey’s multiple comparisons test; asterisks indicate results of post hoc test, and asterisks above bars indicate comparison to dimethyl sulfoxide (DMSO)–treated sample]. (C) Immunoblot of NOXA expression in 1205Lu WT cells treated with an sgRNA targeting the NOXA gene PMAIP1 (sgNOXA) or nontargeting control (sgCTRL). Representative of two independent experiments. (D) Cell death induction in the cell lines described in (C) 18 hours after 3p-RNA transfection (n = 3, Student’s t test). (E) NOXA expression assessed by Immunoblot in 1205Lu WT cells treated with 3p-RNA or poly(I:C) for 6 hours. Representative of two independent experiments. (F) Cell death induction by poly(I:C) (1 μg/ml) or 3p-RNA (160 nM) with or without CHX (10 μg/ml) and with or without transfection reagent (n = 2 to 4, one-way ANOVA, followed by Sidak’s multiple comparisons test). Uncomplexed poly(I:C) was added 6 hours before treatment with CHX. CHX, transfected poly(I:C), or transfected 3p-RNA was added for 12 hours before analysis. (G) qRT-PCR analysis of ACTB (encoding for β-actin), MCL1, and PMAIP1 (encoding for NOXA) expression in WT and RNase L–deficient 1205Lu cells at indicated time points after 3p-RNA treatment (bars represent mean differences in crossing point (Cp) value between mock and 3p-RNA–treated cells ± SEM of three to four independent experiments, Student’s t test; asterisks above bars indicate comparison to untreated control). (H) Immunoblots of IFN-α–primed 1205Lu WT and RNASEL−/− cells stimulated with 3p-RNA for indicated time points or with CHX (10 μg/ml) for 14 hours. Immunoblot shows representative results of three independent experiments. (I) Immunoblot for MCL-1 and NOXA expression of IFN-α–primed 1205Lu WT cells treated with an sgRNA targeting PMAIP1 (sgNOXA) or nontargeting control (sgCTRL) stimulated with 3p-RNA for indicated time points or with CHX (10 μg/ml) for 14 hours (sgCTRL only). Representative results of two independent experiments. (J) Schematic representation of the proposed mechanism of MCL-1 depletion in response to RNase L activation. Unless otherwise stated, means + SEM of independent experiments are shown; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
” data-icon-position=”” data-hide-link-title=”0″>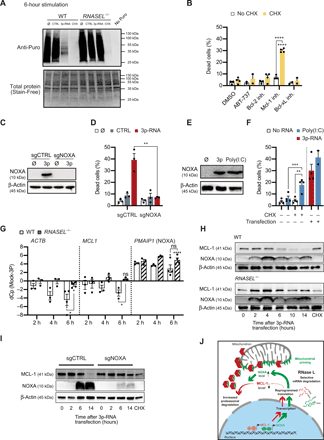
(A) Measurement of puromycin (Puro) incorporation detected by anti–puromycin (Puro) blot (top image) and total protein (Stain-Free technology, bottom image) in IFN-α–primed 1205Lu WT or RNASEL−/− cells treated with CTRL, 3p-RNA (both 160 nM), or CHX (10 μg/ml) for 6 hours. Representative of two independent experiments. (B) Cell death induction in 1205Lu WT cells 24 hours after stimulation with small-molecule inhibitors of BCL2 family proteins (all used at 1 μM) in the presence or absence of CHX (1 μg/ml) [n = 2 to 4, two-way ANOVA, followed by Tukey’s multiple comparisons test; asterisks indicate results of post hoc test, and asterisks above bars indicate comparison to dimethyl sulfoxide (DMSO)–treated sample]. (C) Immunoblot of NOXA expression in 1205Lu WT cells treated with an sgRNA targeting the NOXA gene PMAIP1 (sgNOXA) or nontargeting control (sgCTRL). Representative of two independent experiments. (D) Cell death induction in the cell lines described in (C) 18 hours after 3p-RNA transfection (n = 3, Student’s t test). (E) NOXA expression assessed by Immunoblot in 1205Lu WT cells treated with 3p-RNA or poly(I:C) for 6 hours. Representative of two independent experiments. (F) Cell death induction by poly(I:C) (1 μg/ml) or 3p-RNA (160 nM) with or without CHX (10 μg/ml) and with or without transfection reagent (n = 2 to 4, one-way ANOVA, followed by Sidak’s multiple comparisons test). Uncomplexed poly(I:C) was added 6 hours before treatment with CHX. CHX, transfected poly(I:C), or transfected 3p-RNA was added for 12 hours before analysis. (G) qRT-PCR analysis of ACTB (encoding for β-actin), MCL1, and PMAIP1 (encoding for NOXA) expression in WT and RNase L–deficient 1205Lu cells at indicated time points after 3p-RNA treatment (bars represent mean differences in crossing point (Cp) value between mock and 3p-RNA–treated cells ± SEM of three to four independent experiments, Student’s t test; asterisks above bars indicate comparison to untreated control). (H) Immunoblots of IFN-α–primed 1205Lu WT and RNASEL−/− cells stimulated with 3p-RNA for indicated time points or with CHX (10 μg/ml) for 14 hours. Immunoblot shows representative results of three independent experiments. (I) Immunoblot for MCL-1 and NOXA expression of IFN-α–primed 1205Lu WT cells treated with an sgRNA targeting PMAIP1 (sgNOXA) or nontargeting control (sgCTRL) stimulated with 3p-RNA for indicated time points or with CHX (10 μg/ml) for 14 hours (sgCTRL only). Representative results of two independent experiments. (J) Schematic representation of the proposed mechanism of MCL-1 depletion in response to RNase L activation. Unless otherwise stated, means + SEM of independent experiments are shown; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Induction of intrinsic apoptosis in many cases critically depends on the inhibition of antiapoptotic proteins of the BCL2 family. We therefore identified the antiapoptotic protein that cells depend on in the setting of translational arrest. To this end, we measured induction of death in cells treated with specific small-molecule inhibitors of BCL2 family proteins with and without inhibition of translation by CHX. Only the combined inhibition of the BCL2 family protein myeloid cell leukemia 1 (MCL-1) with S63845 and translation was able to trigger apoptosis in melanoma cells within 24 hours (Fig. 5B), whereas translational arrest or the MCL-1 inhibitor alone was unable to induce cell death in the same time frame.
The antiapoptotic activity of MCL-1 is described to be mainly regulated by its protein level. The cellular MCL-1 level is tightly regulated by transcription, translation, and proteasomal degradation, with a particularly high protein turnover (42, 43). NOXA is a BH3-only protein that binds MCL-1 and BCL2A1 (44). Upon binding to MCL-1, NOXA leads to its proteasomal degradation (45, 46). Given that NOXA is highly up-regulated by RLR ligands and has been implicated in RLR-induced cell death (10, 47), we investigated its involvement in RLR/RNase L–driven apoptosis by targeting the NOXA gene PMAIP1 using CRISPR-Cas9 (Fig. 5C). Cells treated with a single-guide RNA (sgRNA) against PMAIP1 showed complete resistance toward 3p-RNA–induced cell death at 18 hours after transfection (Fig. 5D). On the other hand, activation of Toll-like receptor 3 (TLR3) with polyinosinic-polycytidylic acid [poly(I:C)] was able to induce NOXA to a similar extent as 3p-RNA (Fig. 5E) but was insufficient to trigger apoptosis by itself (Fig. 5F). In line with our previous observations, however, blocking translation after priming cells with poly(I:C) for 6 hours led to apoptosis (Fig. 5F). These findings illustrate that NOXA induction through pattern recognition receptor (PRR) signaling is crucial but insufficient for cell death induction.
Recently, two studies found the promotion of widespread mRNA turnover to account for the translational shutdown seen in response to dsRNA (48, 49). Some mRNAs involved in antiviral defense (e.g., IFNB1) were described to be partially protected from RNase L–mediated degradation. We hypothesized that this mechanism also played a role in RNase L–driven execution of intrinsic apoptosis. Thus, we reanalyzed the published RNA-seq results from these two studies with regard to pro- and antiapoptotic BCL2 family members. This revealed skewing toward the expression of genes encoding proapoptotic BH3-only proteins at 4 hours after poly(I:C) transfection (fig. S5B). Among the BH3-only family members, NOXA and PUMA mRNAs were the only highly and consistently induced ones, driving the asymmetry between pro- and antiapoptotic gene expression (fig. S5C). In addition, mRNAs of NOXA and PUMA were the only ones significantly up-regulated by poly(I:C) among those of the BH3-only proteins and are less prone to degradation by RNase L [data analyzed from (50)].
Aiming to confirm these results in our model system and to further understand the involvement of RNase L activity in the regulation of MCL-1 and NOXA expression, we studied their expression in WT and RNase L–deficient cells primed with IFN-α during the first hours of 3p-RNA treatment on the mRNA and protein levels [Fig. 5, G and H). The mRNAs of β-actin and MCL-1 were rapidly degraded in an RNase L–dependent fashion within 6 hours after 3p-RNA versus mock treatment, whereas NOXA mRNA was induced rapidly and consistently in WT and RNase L–deficient cells. On the protein level, WT cells showed rapid loss of MCL-1 (Fig. 5H). Cells deficient in RNase L could maintain detectable levels of MCL-1 protein until at least 14 hours after 3p-RNA transfection. This likely corresponds to the intact translational machinery in those cells and therefore the ability to maintain MCL-1 protein production. As expected from our quantitative real-time polymerase chain reaction (qRT-PCR) data, rapid induction of NOXA protein could be observed from 2 hours after treatment with 3p-RNA, which was not affected by RNase L. Treatment with CHX alone did not reduce MCL-1 protein.
Because increased proteasomal degradation of MCL-1 is a central mechanism of its depletion in cells, we analyzed MCL-1 protein levels in cells treated with or without the proteasome inhibitor MG-132 (fig. S5D). Inhibiting the proteasome greatly increased the baseline MCL-1 protein level, illustrating its high and rapid turnover. 3p-RNA–induced decrease in MCL-1 at 10 hours was rescued by adding MG-132 to a level comparable to the baseline expression without the inhibitor. Arresting proteasomal degradation of MCL-1 reduced cell death induction by 3p-RNA at early time points (fig. S5E). In line with these findings, cells treated with an sgRNA targeting NOXA did not show substantial MCL-1 depletion upon 3p-RNA stimulation (Fig. 5I). In conclusion, our results suggest a model in which MCL-1 depletion is facilitated by the cooperative action of RNase L–mediated selective translational arrest and up-regulation of NOXA as a mitochondrial priming step ultimately leading to BAX/BAK-dependent intrinsic apoptosis (Fig. 5J).
DISCUSSION
Here, we describe a molecular mechanism of tumor cell death induction upon cytosolic recognition of dsRNA by PRR. Our findings show that apoptosis induction depends on the concerted action of two receptor families: RIG-I is required for IFN-I– and NOXA-dependent priming, whereas the OAS/RNase L system executes apoptosis in primed cells through translational arrest.
Whereas the OAS/RNase L system and PKR are well recognized for their induction of translational arrest and cell death during viral infections and after transfection of the long dsRNA analog and MDA5 ligand poly(I:C) in cell culture (51–55), 3p-RNA as the bona fide RIG-I ligand has been described to induce cell death through direct downstream RIG-I signaling effects (9–11, 14, 25). The high dependence of cell death induction on intact RIG-I and MAVS in cell lines, also shown in our study, leaves little room for other interpretations. One study suggested that cytokine and cell death induction are uncoupled by analyzing a variety of structured single-stranded RNAs with 5′-triphosphate moieties, but the underlying mechanisms remained elusive and were not assigned to a secondary receptor family (56). In addition, there have been contradictory reports on the role of IFN-I in cell death mediated by RLR ligands (10, 13). Studies published so far relied on small interfering RNA (siRNA)–mediated knockdown of pathway components with two potential pitfalls: (i) counteracting up-regulation of targets via IFN-I feed-forward loop and (ii) studying RNA recognition pathways using dsRNA-based tools like siRNA may carry the risk of off-target effects. To address both, we conducted experiments using stable genetically deleted cell lines. In contrast to earlier publications that highlighted the role of IRF3 in transcription-independent apoptosis in both RLR and cyclic GMP-AMP synthase (cGAS) signaling (15, 16, 57), we found cell death to be entirely independent of IRF3, if provided with exogenous IFN-I. The lack of discrimination between IFN-I priming and effector mechanisms may have disguised an interpretation in accordance with our proposed model.
Although the 3p-RNA used in our study fits in the length preference of OAS1, an unexpected finding was the dependence of OAS1 activation on the 5′-triphosphate moiety of a short dsRNA of 20 base pairs (bp). This feature has been controversially described for OAS1. Whereas one study (58) showed an enhanced OAS1 activity using adenoviral-associated RNAs with a 5′-triphosphate moiety compared with dephosphorylated RNAs, another publication (33) found no differences in activity after 5′-end modification. Isolated biochemical studies using recombinant human OAS1 and the use of OAS1-deficient cells support that the 5′-triphosphate group is important for sensing of RNAs by OAS1, and the observed phenotypes are not due to alternative functions of the 5′-triphosphate moiety–like transfection or stability differences or RNA sequestration by other sensors in the cellular context. Further structural and functional analysis using various RNA species with and without 5′-triphosphate may solve this discrepancy and enhance the understanding of viral RNA recognition by OAS1. Because of the heterogeneity of viral RNAs, it was still elusive whether the same RNA species activate the RLR pathway and OAS proteins. Here, we show that 3p-RNA binds to and activates both RIG-I and OAS1 suggesting that they share ligand requirements.
Our results demonstrate that cell death induction is dependent on BAX/BAK pore formation and caspase activity and therefore uses the intrinsic apoptosis machinery. The most consolidated mechanistic finding of 3p-RNA–induced apoptosis in the literature is the involvement of NOXA in this process. However, the preserved up-regulation of NOXA in RNase L–deficient cells or upon TLR3 stimulation despite the lack of apoptosis induction demonstrated that NOXA’s proapoptotic activity is not sufficient. Rather, we provide evidence that NOXA serves as a second “mitochondrial” priming step that is needed for efficient and fast cell death execution by RNase L via the inhibition of translation. We revealed a functional connection of both pathways in the depletion of the antiapoptotic protein MCL-1. It is also conceivable that other antiapoptotic proteins with rapid turnover are depleted through translational arrest, like BCL2A1, that is also antagonized by NOXA. NOXA up-regulation by TLR3 stimulation and translational inhibition by CHX treatment recapitulated the combined actions of 3p-RNA. Therefore, the priming phase can most likely be substituted by any given PRR signaling inducing NOXA and IFN-I, and the effector pathway can be mimicked by inhibition of cellular translation. This is of particular interest because the main effect of OAS/RNase L and similarly of PKR and IFIT1 is translational arrest, which seems to be the common denominator of antiviral cell death pathways. The selective degradation of mRNAs by RNase L spares mRNAs of essential antiviral immune response genes, e.g., IFN-β (48, 49) and, as shown here, NOXA, which differentiates the RNase L pathway from PKR-mediated translational inhibition and may facilitate the efficient onset of cytokine production and apoptosis despite a global shutdown of translation. Whether the engagement of RNase L, compared with PKR, leads to distinct systemic immunological consequences could be of great interest for tumor immunotherapy.
Although we have used different cell lines in this study to generalize our findings, we cannot exclude that cell type–specific receptor repertoires may introduce variability in sensitivity and mode of action of cell death induction. For example, healthy somatic cells seem to be less susceptible to 3p-RNA–induced apoptosis because of a compensatory BCL-XL up-regulation compared with tumor cells (9). This difference may be exploited by the use of 3p-RNA as anticancer drugs that have been successfully tested in various preclinical cancer models particularly in combination with immune checkpoint inhibition (9, 11, 20, 21, 59, 60). A recent study investigating 3p-RNA treatment in combination with CTLA-4 blockade showed local tumor control by intratumoral 3p-RNA treatment to be largely independent of tumor cell–intrinsic RIG-I signaling (59). It is tempting to speculate that tumor cell–intrinsic RNase L signaling rather than RIG-I–mediated cytokine release alone is key for success of immunotherapy using 3p-RNA.
Here, we demonstrate that cytokine release and cell death induction are two separable events downstream of cytoplasmic 3p-RNA recognition. Whereas RLR signaling leads to transcriptional priming of the OAS/RNase L pathway and a second mitochondrial priming step via NOXA, the execution of cell death is dependent on translational arrest triggered by activity of the OAS/RNase L system. This two-step mechanism consisting of priming and effector phases reminiscent of NLR family pyrin domain containing 3 (NLRP3) inflammasome activation appears to be a common mechanism in innate immunity allowing the cell to either cope or perish depending on the insult taken. In addition, as both pathways are “druggable,” a detailed understanding of the molecular mechanisms may pave the way for the development of novel anticancer drugs inducing immunogenic cell death.
METHODS
Study design
This study explored the molecular signaling mechanisms underlying cell death induction downstream of RIG-I ligand transfection in various tumor cells. Quantitative MS, RNA-seq, gain-of-function and CRISPR-Cas9–based loss-of-function cell culture experiments, and biochemical enzyme assays using purified OAS1 were used to decipher the described priming and effector phases of cell death induction. Sample size was chosen on the basis of empirical knowledge of effect sizes. No blinding or randomization was performed. For all experiments, the number of independent replicates and the statistics applied are indicated in the corresponding figure legend.
Cell culture
The human melanoma cell line 1205Lu was provided by R. Besch (University Hospital, LMU Munich, Germany) and validated by short tandem repeat analysis. Human C8161 and murine B16F10 WT and Ddx58−/− melanoma cell lines were a gift of M. Helms (Sanofi) [described in (60)], and B16.OVA WT and Irf3−/− x Irf7−/− cell lines were a gift of S. Heidegger (School of Medicine, Technical University Munich) and H. Poeck (University Hospital Regensburg) and were described previously (21, 59). All tumor cell lines used were cultivated in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, 2 mM l-glutamine, penicillin (100 U/liter), streptomycin (0.1 mg/ml) (all Gibco, Thermo Fisher Scientific, Karlsruhe, Germany). All cell lines were routinely tested for absence of contamination with mycoplasma by PCR.
Cell stimulation
For cell death assays, 1.2 × 104 cells per well were plated in 96-well plates with or without human IFN-α2a [1000 U/ml; research grade (Miltenyi)] the day before transfection. RNA was transfected using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. RNA (0.1 nmol) was complexed using 2.5 μl of transfection reagent. Cell death induction was analyzed 48 hours after RNA stimulation, if not indicated otherwise.
Immunostimulatory RNAs
3p-RNA was custom-synthesized by Ella Biotech (Munich) or generated by in vitro transcription (IVT) from a dsDNA template (5′-GCGCTATCCAGCTTACGTAGAGCTCT ACGTAAGCTGGATAGCGCTATAGTGAGTCGTATTA-3′) using the HiScribe T7 Quick High Yield RNA Synthesis Kit (New England Biolabs GmbH, Frankfurt, Germany) according to the manufacturer’s protocol for small RNAs. 3p-RNA was purified using the Total RNA Clean-Up and Concentration Kit (Norgen Biotek, Canada), after which the purity was determined by high performance liquid chromatography (LC) using a DNAPac 200 column on a GE Äkta micro system (GE Healthcare). Double-stranded CTRL (5′-GCGCUAUCCAGCUUACGUA-3′) was ordered at Metabion (Munich) and used as transfection control as previously described (10). Synthetic hairpin OH-RNA (5′- GCGCUAUCCAGCUUACGUAGAGCUCUACGUAAGCUGGAUAGCGC-3′) was used to control for 5′-triphosphate–dependent effects. A published OAS1-activating positive-control 3′-ssPy [5′-GGCUUUUGACCUUUAUGCU-3′ (forward) and 5′-GCAUAAAGGUCAAAAGCC-3′ (reverse)] (61) was purchased from Metabion, annealed in a 1:1 molar ratio, and used as positive control for OAS1 activation.
For generation of total RNA from noninfected or from virally infected 1205Lu DDX58 KO cells, total RNA was isolated 24 hours after infection (VSV-WT and VSV-M51R, multiplicity of infection 1; SeV, 40 U/ml). Total RNA was complexed using Lipofectamine RNAiMAX and transfected at 1 μg/ml, if not indicated otherwise.
Dephosphorylation of 3p-RNA was performed by incubating 10 μg of RNA in the presence of 30 U of FastAP Thermosensitive Alkaline Phosphatase (FastAP, Thermo Fisher Scientific) or mock control in the appropriate reaction buffer for 2 hours at 37°C, followed by a heat-inactivation step for 10 min at 75°C. RNA was then column-purified as described above. Dephosphorylation and RNA integrity were confirmed by high-performance LC. The synthetic RNA analogon poly(I:C) (HMW) VacciGrade was purchased from Invivogen.
CRISPR-Cas9–mediated gene KO
1205Lu DDX58, IFIH1, and MAVS KO cells were generated by transient transfection of pCMV-mCherry-Cas9 plasmids, harboring the respective sgRNA, which were provided by V. Hornung (Gene Center, LMU Munich). All other KOs were produced by transient expression of the enhanced SpCas9(1.1) together with the respective sgRNA (table S1) targeting the gene of interest from a 2A-PuroR or 2A-BlastR-modified eSpCas9(1.1) plasmid. pSpCas9(BB)-2A-Puro (PX459) v2.0 (62) and eSpCas9(1.1) (63) were a gift from F. Zhang (Addgene, plasmid nos. 62988 and 71814, respectively). Transfected cells were enriched by puromycin/blasticidin selection for 48 hours, and if indicated, single-cell clones were established by limiting-dilution cultivation. 1205Lu DDX58, IFIH1, and MAVS KO cells were validated by Sanger sequencing, whereas all other KOs were confirmed by next-generation sequencing (Illumina MiSeq) of the target locus as described in (64) and via Western blot. To avoid clonal artifacts, at least three validated clones were pooled before experiments. If indicated, in the figure legends, a nontargeting control sgRNA (sgCTRL) was used.
Measurement of apoptosis and IFN-stimulated genes induction in single and cocultures by flow cytometry
For flow cytometric analysis of apoptosis induction, floating and adherent cells were collected and stained with recombinant annexin A5–allophycocyanin, fluorescein isothiocyanate, or phycoerythrin (1:50, all ImmunoTools) together with Fixable Viability Dye eFluor 780 (1:5000, eBioscience). Caspase activity was assessed by staining with Violet Live Cell Caspase Probe (BD Pharmingen) as the provider suggests. To estimate IFN-I signaling strength by flow cytometry we determined MHC-I expression using Alexa Fluor 488 or Alexa Fluor 647 antihuman human leukocyte antigen-A, -B, -C antibody (1:250, BioLegend) for human or Alexa Fluor 488 anti-mouse H-2Kb antibody (1:250, BioLegend) for mouse cell lines. Data were acquired on a BD LSRFortessa flow cytometer and analyzed using FlowJo software (BD).
A coculture setup was used to investigate cell death induction in RLR signaling–deficient cells. To this end, either WT or KO cells were labeled using Cell Proliferation Dye eFluor 450 (eBioscience) or CellTrace CFSE Cell Proliferation Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions before seeding. Labeled and unlabeled cells were mixed in a 1:1 ratio and seeded at a total of 1.2 × 104 cells per 96 wells. To ensure proper discrimination of cell lines by flow cytometry, labeled and unlabeled cells were cultivated and analyzed as single controls.
Cell viability assay
Cellular viability was quantified by CellTiter-Blue (CTB) Cell Viability Assay (Promega). The CTB reagent was added to cell culture medium at 20% (v/v) and incubated for 2 to 4 hours, and fluorescence was recorded at 560 excitation/590 emission on a Mithras LB 940 multimode plate reader (Berthold). Viability was calculated from the following formula: (Fluorescence sample − Fluorescence medium control)/(Fluorescence untreated control − Fluorescence medium control) × 100%.
Quantification of IFN-β and CXCL10
For quantification of IFN-β or CXCL10 protein levels, cell-free supernatant was subjected to enzyme-linked immunosorbent assay (ELISA) using Human IFN-beta DuoSet ELISA, Mouse IFN-beta DuoSet ELISA, or Human CXCL10/IP-10 DuoSet ELISA (all R&D Systems) according to the manufacturer’s instructions.
Inhibitors and blocking antibodies
Small-molecule inhibitors and blocking antibodies were added to cell culture medium 1 hour before stimulation. Inhibitors were used at the following concentrations, if not otherwise indicated: z-VAD, 20 μM (+) or 80 μM (++) (Invivogen); ABT-737, 1 μM (Adooq Biosciences); venetoclax, 1 μM (Selleckchem); S63845, 1 μM (Selleckchem); A-1155463, 1 μM (Adooq Biosciences); and MG-132, 10 μM (Selleckchem). To block IFNAR signaling, Ultra-LEAF Purified antimouse IFNAR1 antibody (MAR1-5A3) or Ultra-LEAF Purified mouse immunoglobulin G1, κ isotype control antibody (both 5 μg/ml; both BioLegend) was used.
RNA isolation and qRT-PCR
Total cellular RNA was isolated using the PeqGOLD Total RNA Kit (VWR International GmbH, Darmstadt, Germany) according to the manufacturer’s recommendations. Complementary DNA (cDNA) was synthesized using a SuperScript II reverse transcription kit (InvivoGen, Toulouse, France) using oligo(dT) primers. qRT-PCR was performed on a Roche LightCycler 480 II system (Hofmann–La Roche, Basel, Switzerland) using the Biozym Probe qPCR Kit (Biozym, Hessisch Oldendorf, Germany) in a total reaction volume of 10 μl per sample. All samples were run in duplicate and referenced to ACTB. Relative expression analysis was performed using the delta Cq method. Primers and hydrolysis probes were selected via the Roche Universal Probe Library assay design center.
RNA degradation and sequencing
RNA concentration and integrity were measured with the RNA 6000 Pico Kit (Agilent) on an Agilent Bioanalyzer 2100. For RNA-seq, contaminating DNA was removed with the RapidOut DNA Removal Kit (Thermo Fisher Scientific). mRNA enrichment and cDNA library preparation were done using Poly(A) mRNA Magnetic Isolation Module and NEBNext Ultra II Directional RNA Library Prep Kit (both New England Biolabs GmBH) according to the manufacturer’s instructions. Paired-end sequencing (2 × 75 bp) was performed at the Dr. von Hauner Children’s Hospital next generation sequencing facility using a NextSeq 500 system with 24 samples per HighOutput cartridge (Illumina).
Bioinformatic analysis
Quality of sequencing reads was assessed using fastQC (www.bioinformatics. babraham.ac.uk/projects/fastqc). Reads were mapped against the human genome (hg38) and human rRNA sequences with ContextMap version 2.7.9 (65) using Burrows-Wheeler Alignment tool (66) as internal short read aligner and allowing at most four mismatches per read. Read counts per gene were determined from mapped RNA-seq reads using featureCounts (67) using ENSEMBL gene annotations (version 84). Differential gene expression analysis was performed on gene read counts using limma for all genes with an average of 25 reads per sample (68). P values were adjusted for multiple testing using the method by Benjamini and Hochberg (69), and genes with an adjusted P value of <0.001 were considered significantly differentially expressed. The RNA-seq analysis workflow was developed and executed using the workflow management system Watchdog (70). Functional enrichment analysis for GO:MF terms was performed for significantly up-regulated genes (adjusted P value of <0.001) using clusterProfiler (71) and org.Hs.eg.db (v3.7.0) (72). All genes included in the differential gene expression analysis were used as background. Terms with adjusted P < 0.001 were considered significant.
3p-RNA affinity purification and MS
Affinity purification
A total of 3 × 106 1205Lu cells were plated in 14-cm dishes until they reached near confluence and stimulated with IFN-α2a (2000 U/ml) for 16 hours. Subsequently, cells were trypsinized, transferred into a microcentrifuge tube, washed twice with phosphate-buffered saline (PBS) and lysed in 1 ml of RNA-immunoprecipitation buffer [20 mM tris HCl (pH 7.5), 50 mM NaCl, 0.25% NP-40, 1.5 mM MgCl2, 1 mM NaF, protease inhibitor cocktail (P8340, Roche), RiboLock RNase Inhibitor (Thermo Fisher Scientific)] for 30 min on ice. Insoluble material was pelleted at 16,000g for 20 min at 4°C, and cleared lysate was stored at −20°C. Biotinylated 3p-RNA was generated by IVT using the HiScribe T7 High Yield RNA Synthesis Kit (New England Biolabs GmbH) according to the manufacturer’s protocol for small RNAs. Uridine 5′-triphosphates (UTPs; 2.5 mM) were exchanged with Biotin-16-UTP (Biozym). IVT products were column-purified as described above. Hydrophilic magnetic streptavidin beads (60 μl; New England Biolabs GmbH) per condition were incubated with an excess of biotin-labeled 3p-RNA (3p-RNA-biot) in PBS supplemented with RNase inhibitor (RiboLock, Thermo Fisher Scientific) for 2 hours at 4°C. Beads were washed three times with ice-cold PBS, resuspended in 1 mg freshly thawed cleared protein lysate in RNA-IP buffer, and rotated for 3 hours at 4°C. After four times rigorous washing in RNA-IP buffer, beads were resuspended in Laemmli buffer and heated for 5 min at 95°C.
Sample preparation for MS
Co-immunoprecipitation samples in reducing Laemmli buffer were subjected to a modified single-pot solid phase–enhanced sample preparation (SP3) protocol (73). Briefly, 10 μl of a bead slurry of Sera-Mag SpeedBeads A and B (4 μg/μl; GE Healthcare, USA) was added to the samples. Protein binding to the magnetic beads was achieved by adding acetonitrile (ACN) to a final volume of 70% (v/v) and mixing at 1200 rpm at 24°C for 30 min in a thermomixer (Eppendorf, Germany). Magnetic beads were retained in a DynaMag-2 magnetic rack (Thermo Fisher Scientific, USA), and the supernatant was discarded. Cysteines were alkylated by adding 25 μl of 80 mM iodoactemamide (Sigma-Aldrich, Germany) and incubation at 1200 rpm at 24°C for 30 min in the dark in a thermomixer. The reaction was quenched by adding 3 μl of 200 mM dithiothreitol (Biozol, Germany). Protein binding to the beads was repeated in 70% (v/v) ACN for 30 min. After removing the solvent, beads were washed twice in 200 μl of 70% (v/v) ethanol and twice in 180 μl of 100% (v/v) ACN. Next, 250 ng of LysC and 250 ng of trypsin (Promega, Germany) were added in 20 μl of 50 mM ammonium bicarbonate (Sigma-Aldrich, Germany). The protein digestion was performed for 16 hours at room temperature. Samples were acidified with formic acid to a final concentration of 1% (v/v) and placed in the magnetic rack. The supernatants were transferred into fresh 0.5-ml Protein LoBind tubes (Eppendorf, Germany). A volume of 20 μl of 2% (v/v) dimethyl sulfoxide was added to the beads and subjected to sonication for 30 s in a water bath. Tubes were placed in the magnetic rack, and the supernatants were transferred to the same tubes. The samples were dried in a vacuum centrifuge and dissolved in 20 μl of 0.1% formic acid.
LC–tandem MS analysis
Samples were analyzed by LC–tandem MS (LC-MS/MS) for relative label free protein quantification. A volume of 10 μl per sample was separated on a nanoLC system (EASY-nLC 1200, Thermo Fisher Scientific, USA) using an in-house packed C18 column (30 cm by 75 μm internal diameter; ReproSil-Pur 120 C18-AQ, 1.9 μm; Dr. Maisch GmbH, Germany) with a binary gradient of water (A) and ACN (B) containing 0.1% formic acid at 50°C column temperature and a flow rate of 250 nl/min (gradient: 0 min, 2.4% B; 2 min, 4.8% B; 92 min, 24% B; 112 min, 35.2% B; and 121 min, 60% B).
The nanoLC was coupled online via a nanospray flex ion source (Proxeon—part of Thermo Fisher Scientific, USA) equipped with a PRSO-V2 column oven (Sonation, Germany) to a Q-Exactive HF mass spectrometer (Thermo Fisher Scientific, USA). Full MS spectra were acquired at a resolution of 120,000. The top 15 peptide ions were chosen for higher-energy C-trap dissociation with a normalized collision energy of 26%. Fragment ion spectra were acquired at a resolution of 15,000. A dynamic exclusion of 120 s was used for peptide fragmentation.
Data analysis and LFQ
The raw data were analyzed by the software Maxquant (maxquant.org, Max-Planck Institute Munich) version 1.5.5.1 (74). The MS data were searched against a reviewed canonical fasta database of Homo sapiens from UniProt (downloaded: 9 January 2018, 20,243 entries). Trypsin was defined as protease. Two missed cleavages were allowed for the database search. The option first search was used to recalibrate the peptide masses within a window of 20 parts per million (ppm). For the main search peptide and peptide, fragment mass tolerances were set to 4.5 and 20 ppm, respectively. Carbamidomethylation of cysteine was defined as static modification. Acetylation of the protein N terminus and oxidation of methionine were set as variable modifications. The false discovery rate for both peptides and proteins were adjusted to less than 1%. LFQ of proteins required at least two ratio counts of razor peptides. Only unique and razor peptides were used for quantification. The option “match between runs” was enabled with a matching time of 1 min. Samples were normalized separately for each batch of biological replicates.
LFQ protein ratios were calculated between the two groups (3p-RNA/Bead, pulldown from stimulated lysates; phosphatase-treated 3p-RNA/Bead, pulldown from stimulated lysates) separately for each biological replicate. After log2 transformation, a one-sample t test was used to evaluate statistically significant changed abundances of proteins between the different sample groups. A P value less than 0.05 was set as significance threshold.
Immunoblotting
Cells were lysed by resuspending pelleted cells in ice-cold lysis buffer containing 50 mM tris-HCl (pH 7.8), 137 mM NaCl, 0.5 mM EDTA, 1 mM sodium orthovanadate, 10% (w/v) glycerol, 1% (w/v) NP-40 supplemented with a protease inhibitor cocktail (P8340, Sigma-Aldrich), followed by 10-min incubation on ice. Insoluble material was pelleted by 16,000g, 4°C, 10 min. Protein concentration of cleared lysate was determined using DC Protein Assay (Bio-Rad), and 35 μg of protein of each sample was separated by 10, 12, or 4 to 20% denaturing SDS–polyacrylamide gel electrophoresis [Mini-PROTEAN TGX Stain-Free Protein Gels (Bio-Rad)], followed by Western blotting on polyvinylidene difluoride membrane using the TransBlot Turbo System (Bio-Rad). Blots were incubated with the primary antibodies to OAS1 (D1W3A, 1:1000; Cell Signaling Technology), OAS2 [polyclonal (19279-1-AP), 1:1000; Proteintech], OAS3 [polyclonal (GTX118059), 1:1000; Genetex], PKR (D7F7, 1:1000; Cell Signaling Technology), BAX [polyclonal (#2772), 1:1000; Cell Signaling Technology], BAK (D4E4, 1:1000; Cell Signaling Technology), Mcl-1 (clone 22, 1:200; Santa Cruz Biotechnology), NOXA (clone 114C307, 1:200; Santa Cruz Biotechnology), β-actin–horseradish peroxidase (C4, 1:2000; Santa Cruz Biotechnology), or glyceraldehyde-3-phosphate dehydrogenase [polyclonal (FL-335), 1:1000; Santa Cruz Biotechnology].
For measurement of translational activity by immunoblot, cells were pulsed with puromycin (1 μg/ml) 30 min before lysis. Total protein was isolated as described above, and 20 μg of protein was separated using 4 to 20% Mini-PROTEAN TGX Stain-Free Protein Gels (Bio-Rad). Total protein load was analyzed by Stain-Free technology on a ChemiDoc Imaging System (Bio-Rad). Puromycin incorporation was assessed using an antipuromycin antibody (12D10, 1:10000; Merck).
OAS1 activity assay
OAS1 recombinant protein expression and purification
Human OAS1 p46 full length was cloned into a modified pET28 vector with an N-terminal His–small ubiquitin-related modifier 1 (SUMO1) tag. Escherichia coli BL21 Rosetta (DE3) cells were cultured in 3 liters of Terrific Broth media until reaching an optical density at 600 nm of 1.0 to 2.0, and protein production was induced at 18°C with 0.4 mM isopropyl-β-d-thiogalactopyranoside for 16 hours. Cell pellets were resuspended in lysis buffer [20 mM Hepes (pH 7.5), 400 mM NaCl, 30 mM imidazole, 10% glycerol, and 1 mM β-mercaptoethanol] and disrupted by sonication. Recombinant OAS1 protein was purified over nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography, and the His-SUMO1 tag was subsequently removed by sentrin-specific protease 2 protease cleavage at 4°C, followed by overnight dialysis against 20 mM Hepes (pH 7.5), 300 mM NaCl, and 1 mM β-mercaptoethanol. Digested protein was separated from His-SUMO1 and protease by a second Ni-NTA affinity chromatography step and further purified by a Superdex 75 16/60 size exclusion chromatography column (GE Healthcare) in 20 mM Hepes (pH 7.5), 250 mM NaCl, and 1 mM tris(2-carboxyethyl)phosphine. Purified OAS1 was concentrated up to 2 to 3 mg/ml, flash-frozen in liquid nitrogen, and stored at −80°C.
Chromogenic assay of OAS1 activity
OAS1 activation by various RNA ligands was analyzed by monitoring pyrophosphate production as a reaction by-product of 2-5A synthesis, as previously described (33). Briefly, OAS1 (300 nM) was incubated with RNA (300 nM) in 25 mM tris (pH 7.5), 10 mM NaCl, 7 mM MgCl2, 1 mM dithiothreitol, and 2 mM adenosine triphosphate at 37°C and a total volume of 150 μl. Aliquots of 10 μl were taken from the reaction over a 0- to 120-min time course and immediately quenched by adding directly to the wells of a 96-well plate predispensed with 2.5 μl of 250 mM EDTA (pH 8.0). To detect pyrophosphate, 10 μl of 2.5% (w/v) ammonium molybdate in 2.5 M H2SO4 and 10 μl of 0.5 M β-mercaptoethanol were added to each well, and the volume was brought to a total of 100 μl with water. Absorbance at 580 nm was measured using a Tecan Infinite M1000 plate reader. Background control (lacking both OAS1 and RNA) was subtracted from the measured values. Experiments were performed as at least three independent assays.
xCELLigence
The kinetic analysis of cell death induction was measured by cellular impedance using an xCELLigence real-time cell analyzer (ACEA Bioscience, San Diego, USA) according to the manufacturer’s protocol. Wells of an E-Plate 96 PET were equilibrated with 100 μl of cell medium containing IFN-α2a (1000 U/ml) for prestimulating conditions. After machine equilibration, 12,000 cells per well were added in 50 μl of medium. Sixteen hours later, RNA was transfected as described above. Impedance was measured for at least 48 hours every 15 min.
Inducible overexpression
OAS1 p46, OAS2, and OAS3 were amplified from cDNA derived from 1205Lu WT cells and cloned into the pLVX-Tight-Puro vector (Clontech). For lentivirus production, human embryonic kidney–293T cells were transfected with pCMV-VSV-G, pCMV-μR8.2, and pLVX-Tight-Puro (containing the gene of interest) or pLVX-Tet-On Advanced in a 1:1:2 ratio. After 48 hours, the supernatant was harvested and passed through a 0.45-μm filter. For transduction of target cells, 1 ml of 800-μl pLVX-Tight-Puro and 200-μl pLVX-Tet-On Advanced supernatant was used per well of a six-well plate. Forty-eight hours after transduction, target cells expressing both constructs were selected using puromycin (2 μg/ml)– and G418 (2 mg/ml)–containing medium.
Measurement of translational activity by SUnSET
Global translational activity was measured by SUnSET (41). Cells were seeded at 2 × 105 per well in six-well plates 1 day before treatment. Transfection of stimulatory RNA was performed in 1 ml of culture medium at 160 nM. Twenty-four hours after RNA treatment, all experimental conditions were pulsed with puromycin (1 μg/ml) in complete medium for 30 min, followed by a chase in puromycin-free medium for 1 hour to facilitate detection of labeled proteins at the cell surface. For flow cytometric analysis, cells were detached by scraping, washed once, and stained with anti–puromycin-Alexa Fluor 647 antibody (12D10, 1:200; Merck).
Quantification and statistical analysis
Gaussian distribution of datasets was assumed. The statistical details of experiments are stated in the corresponding figure legends. If not indicated otherwise, two-sided statistical tests were used, and data represent means + SEM. Analyses were performed with GraphPad Prism (version 8.2.0) and Excel (Microsoft Excel for Mac version 16.27).
Acknowledgments: We thank V. Hornung (Gene Center and Department of Biochemistry, LMU Munich) for discussions and input, N. Schmacke (Gene Center and Department of Biochemistry, LMU Munich) for supporting us with amplicon-based next-generation sequencing of KO cell lines, and Life Science editors for editing support. Cytometry data were obtained in the Core Facility Flow Cytometry of the University Hospital, LMU Munich. Funding: This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (DFG project 442265435 to L.M.K. and project 369799452-TRR237 to L.M.K., K.-P.H., and S.R.); Friedrich-Baur-Stiftung Reg.-Nr. 48/17 to L.M.K.; the Else Kröner-Fresenius-Stiftung 2017_A50 to L.M.K.; the international doctoral program “iTarget: Immunotargeting of cancer” funded by the Elite Network of Bavaria (to M.S., L.M.K., K.-P.H., and S.E.); the Marie-Sklodowska-Curie Training Network for the Immunotherapy of Cancer (IMMUTRAIN, grant number 641549) funded by the H2020 program of the European Union (to S.E., L.M.K., K.-P.H., and M.S.); by the DFG project 329628492–SFB 1321 to M.S., project 315563603 to M.S. and P.D., project 391217598 and 404450088 to S.R., and FR2938/10-1 to C.C.F.; by Cancer Research Institute/Eugene V. Weissman Fellow to C.C.d.O.M.; and by Munich Cluster for Systems Neurology (EXC 2145 SyNergy, ID 390857198) to S.F.L. Author contributions: Conceptualization, D.F.R.B., S.R., K.-P.H., P.D., M.S., and L.M.K. Methodology, D.F.R.B., S.F., C.C.d.O.M., S.A.M., M.R., and L.M.K. Formal analysis, D.F.R.B., S.F., C.C.d.O.M., S.A.M., M.K., and C.C.F. Investigation, D.F.R.B., S.F., C.C.d.O.M., S.A.M., M.K., P.M., M.R., C.H., L.K., and L.M.K. Resources, S.A.M., S.F.L., S.E., and S.R. Writing—original draft, D.F.R.B. and L.M.K. Writing—review and editing, all authors. Visualization, D.F.R.B., S.F., C.C.d.O.M., M.K., C.C.F., and L.M.K. Supervision, K.-P.H., P.D., M.S., and L.M.K. Project administration, P.D., M.S., and L.M.K. Funding acquisition, S.E., S.R., K.-P.H., M.S., and L.M.K. Competing interests: The authors declare that they have no competing interests. Data and materials availability: RNA-seq data can be accessed through the Gene Expression Omnibus (GEO) under the accession number GSE150658. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, L.M.K. (lars.koenig{at}med.uni-muenchen.de). pSpCas9(BB)-2A-Puro (PX459) V2.0 and eSpCas9(1.1) are available from Addgene under a material transfer agreement.
- Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works