INTRODUCTION
The human body is constitutively exposed to various environmental stimuli, and these stimuli are diverse and highly variable with time and location. As the most external organ, the skin is primarily responsible for the protection and appropriate recognition of danger signals in this context (1, 2), but it has also established tolerance mechanisms to adapt to these various challenges from the environment without unwanted inflammation that would promote disease (3, 4). For example, failure of immune tolerance mechanisms results in classic allergic responses to specific antigens, and common human inflammatory skin disorders such as psoriasis, atopic dermatitis, and acne reflect dysfunction of tolerance to innate immune stimuli. However, despite the importance of innate immune tolerance to overall immune homeostasis and its potential relevance to multiple human diseases, the mechanisms which confer innate tolerance of the epidermis to diverse environmental stimuli remain incompletely understood.
Microbial products such as those that trigger activation of Toll-like receptors (TLRs) are a classic example of environmental stimuli that are a potent trigger of inflammation in immunocytes but are well tolerated by keratinocytes in the epidermis. Instead of triggering an immune defense response, the epidermis provides a large surface for positive interactions with microbes and has established symbiotic relationships with some members of the skin microbiome (5, 6). Subsequent to the communication between microbes and the epidermis, these epithelial cells then closely interact with classical immunocytes to shape the overall immune response (7–9). Therefore, understanding how keratinocytes interact with external environmental triggers is an important goal that can enable improved therapy of immune disorders.
A common mechanism for environmental control of cell function is through epigenetic control of gene expression. An important class of molecules involved in epigenetic control is the short-chain fatty acids (SCFAs) that can increase acetylation of histones through inhibition of histone deacetylases (HDACs). SCFAs are generated from bacteria under anaerobic conditions (10) and can accumulate in hair follicles due to fermentation by Cutibacterium acnes (11). Although SCFAs such as butyrate have been known to inhibit inflammatory responses by bone marrow–derived immunocytes (10, 12), an important clue to understand innate immune tolerance of the skin came with the observation that SCFAs increase inflammatory responses by keratinocytes to TLR ligands. Inhibition of HDAC8 and HDAC9 by SCFAs produced by C. acnes amplified inflammatory responses in keratinocytes (13). These observations suggested that microbes could inhibit HDACs and that HDACs are a mechanism through which keratinocytes can tolerate exposure to the varied TLR ligands. This study confirms this hypothesis and provides a detailed understanding of mechanisms by which epigenetic events drive innate immune tolerance in the skin. Our findings suggest that the activity of HDAC8 and HDAC9 in keratinocytes is a central regulator of innate immune tolerance.
RESULTS
HDAC8 and HDAC9 inhibit the inflammatory response to TLR ligands and expression of MAP2K3 in keratinocytes
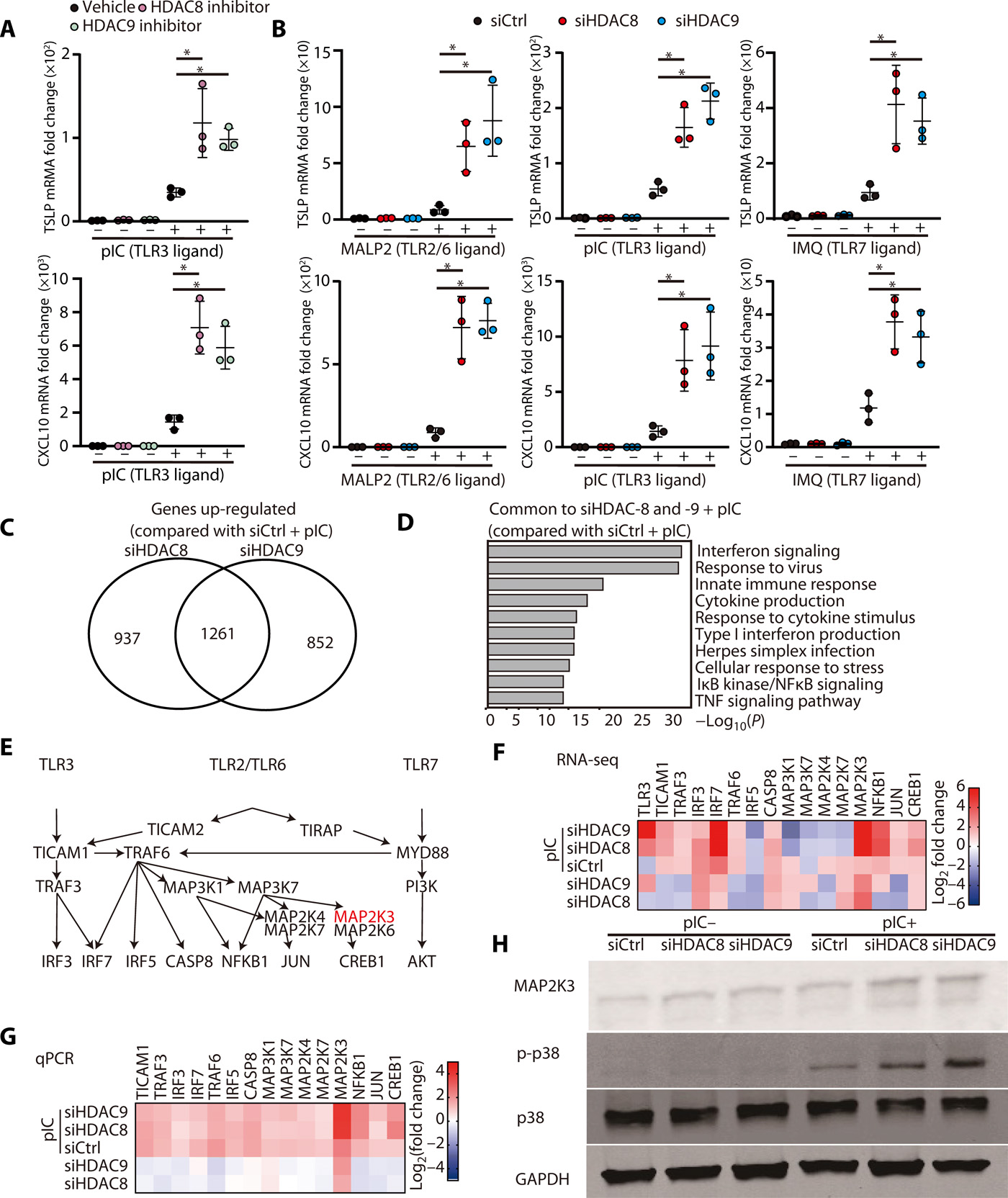
The capacity of microbes to promote inflammation by inhibition of histone deacetylation (13) has demonstrated the need to better understand how HDACs may maintain tolerance at the skin surface to innate stimuli of inflammation such as TLR ligands. Inhibition of HDAC8/9, but not HDAC1 to HDAC7, was previously observed to result in greatly increased cytokine expression in the skin in response to microbial products (13). Addition of selective chemical inhibitors for HDAC8 and HDAC9 to primary cultures of normal human keratinocytes confirmed these prior observations and increased expression of TSLP and CXCL10 in the presence of a TLR3 ligand (Fig. 1A). Targeting of HDAC8 or HDAC9 by RNA silencing of normal human keratinocytes confirmed this result and also increased expression of TSLP and CXCL10 when cells were exposed to ligands for TLR3, TLR2/6, and TLR7 (Fig. 1B). Because no change in cytokine expression was observed after inhibition of HDACs without a TLR stimulus, we hypothesized that HDAC8 and HDAC9 may act to suppress downstream signaling in keratinocytes that was triggered by TLRs.
(A) nHEKs were pretreated for 1 hour with PCI34051 (10 μM) or TMP269 (1 μM) as HDAC8 or HDAC9 inhibitors, respectively, and then cultured for 4 hours with poly I:C (1 μg/ml). mRNA expression of TSLP or CXCL10 was measured by qPCR (N = 3; one-way ANOVA). (B) nHEKs were treated for 24 hours with silencing RNAs to HDAC8 or HDAC9 or control scrambled siRNA and then cultured for 3 hours with MALP2 (200 ng/ml) or poly I:C (1 μg/ml) or for 4 hours with IMQ (30 μg/ml). mRNA expression of TSLP or CXCL10 was measured by qPCR (N = 3; one-way ANOVA). (C) Total RNA-seq after silencing of HDAC8 or HDAC9 and the addition of poly I:C compared with control siRNA and poly I:C. (D) Gene ontology analysis of genes induced by poly I:C after HDAC8/9 silencing compared with genes induced by poly I:C after control siRNA. (E) Illustration of signaling pathways for TLR3, TLR2/6, and TLR7. (F) Heatmap of TLR signaling–related genes that increased after HDAC8/9 silencing as determined by RNA-seq and (G) validated by qPCR. (H) Immunoblotting for phospho-p38MAPK, p38MAPK, MAP2K3, or GAPDH from nHEKs treated with poly I:C after HDAC8/9 silencing. Results are expressed as the means ± SD. *P < 0.05. Data are representative of three independent experiments.
” data-icon-position=”” data-hide-link-title=”0″>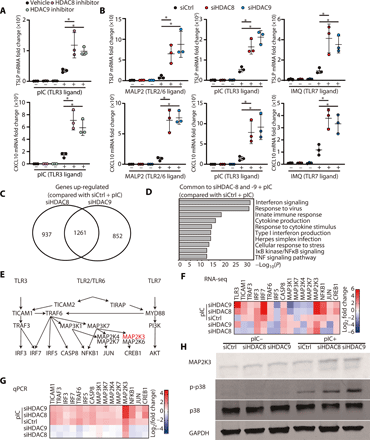
(A) nHEKs were pretreated for 1 hour with PCI34051 (10 μM) or TMP269 (1 μM) as HDAC8 or HDAC9 inhibitors, respectively, and then cultured for 4 hours with poly I:C (1 μg/ml). mRNA expression of TSLP or CXCL10 was measured by qPCR (N = 3; one-way ANOVA). (B) nHEKs were treated for 24 hours with silencing RNAs to HDAC8 or HDAC9 or control scrambled siRNA and then cultured for 3 hours with MALP2 (200 ng/ml) or poly I:C (1 μg/ml) or for 4 hours with IMQ (30 μg/ml). mRNA expression of TSLP or CXCL10 was measured by qPCR (N = 3; one-way ANOVA). (C) Total RNA-seq after silencing of HDAC8 or HDAC9 and the addition of poly I:C compared with control siRNA and poly I:C. (D) Gene ontology analysis of genes induced by poly I:C after HDAC8/9 silencing compared with genes induced by poly I:C after control siRNA. (E) Illustration of signaling pathways for TLR3, TLR2/6, and TLR7. (F) Heatmap of TLR signaling–related genes that increased after HDAC8/9 silencing as determined by RNA-seq and (G) validated by qPCR. (H) Immunoblotting for phospho-p38MAPK, p38MAPK, MAP2K3, or GAPDH from nHEKs treated with poly I:C after HDAC8/9 silencing. Results are expressed as the means ± SD. *P < 0.05. Data are representative of three independent experiments.
To understand the mechanism by which the activity of HDAC8 and HDAC9 suppresses cytokine expression in response to TLR ligands, RNA sequencing (RNA-seq) of keratinocytes was performed. We identified 1261 genes that were commonly up-regulated when HDAC8 or HDAC9 were silenced and also exposed to polyinosinic:polycytidylic acid (poly I:C) (Fig. 1C). The gene sets that increased after silencing of HDAC8 or HDAC9 in the presence of a TLR3 ligand were primarily associated with inflammatory responses (Fig. 1D). RNA-seq of cultured normal human keratinocytes did not detect increased expression of inflammatory genes after silencing of HDAC8/9 without the addition of poly I:C (fig. S1, A and B). These data supported the conclusion that HDAC8/9 does not act directly on the expression of inflammatory genes and provided candidate genes that may mediate the suppressive effect.
TLR2/6, TLR3, and TLR7 have both distinct and overlapping canonical signaling pathways that promote cytokine expression (Fig. 1E). Interrogation of the RNA-seq results identified dual specificity mitogen-activated protein kinase kinase 3 (MAP2K3) and IRF3 (interferon regulating factor 3) from among this signaling cascade as highly induced by silencing of either HDAC8 or HDAC9 (Fig. 1F). Independent reverse transcription quantitative polymerase chain reaction (RT-qPCR) validated that MAP2K3 was increased from among these genes (Fig. 1G), and therefore, we focused analysis on the activity of this important kinase. The function of MAP2K3 was confirmed to be increased because the substrate of this kinase (p38MAPK) showed increased phosphorylation after silencing of HDAC8 and HDAC9 and stimulation by poly I:C (Fig. 1H and fig. S2). These observations suggested that HDAC8 or HDAC9 inhibits MAP2K3 expression and function. The convergence of TLR signaling through this key kinase could explain the effects of HDAC inhibition on inflammatory cytokines.
HDAC8 and HDAC9 interact with SSRP1 and SUPT16H to influence keratinocyte cytokine and MAP2K3 expression
Through assembly into protein complexes and deacetylation at specific histone marks, HDACs are powerful regulators of expression for many target genes (13–21). Understanding the components of these protein complexes is important to elucidate the molecular mechanism for gene regulation by HDACs. To clarify this regulatory mechanism for HDAC8 and HDAC9 in human keratinocytes, immunoprecipitation and subsequent mass spectrometry analysis were performed to identify proteins that associate with HDAC8 and HDAC9. Specificity of anti-HDAC8 and anti-HDAC9 relative to the isotype control antibody was verified by Western blot (fig. S1C). Among known nucleosome proteins, a total of 34 proteins were observed associated with both HDAC8 and HDAC9 (Fig. 2, A and B). In particular, HDAC8 and HDAC9 were both found to be associated with the SSRP1 and SUPT16H proteins, components of the facilitates chromatin transcription (FACT) complex that is known to act as a major regulator of transcription through effects on elongation (22, 23).
(A) Proteins identified by mass spectrometry of nHEK extracts enriched by immunoprecipitation with anti-HDAC8 or anti-HDAC9 compared with control IgG. (B) The common enriched nucleosome proteins precipitated by anti-HDAC8 and anti-HDAC9. (C) Transcriptional elongation FACT proteins SSRP1 and SUPT16H that were identified in (B) were silenced in nHEKs, and then HDAC activity was chemically inhibited by butyrate treatment (2 mM) for 1 hour. nHEKs were subsequently cultured with or without poly I:C (1 μg/ml) for 4 hours, and gene expression was measured by qPCR (N = 3; one-way ANOVA). (D) MAP2K3 transcriptional elongation assay. The transcriptional elongation rate of MAP2K3 was measured after SSRP1 or SUPT16H silencing with or without butyrate treatment (2 mM) for 1 hour. Data show the relative abundance of pulse-labeled RNA for the indicated genes at the indicated times after removal of the transcriptional inhibitor DRB (N = 3; one-way ANOVA). The results are expressed as the means ± SD. *P < 0.05. Data are representative of three independent experiments.
” data-icon-position=”” data-hide-link-title=”0″>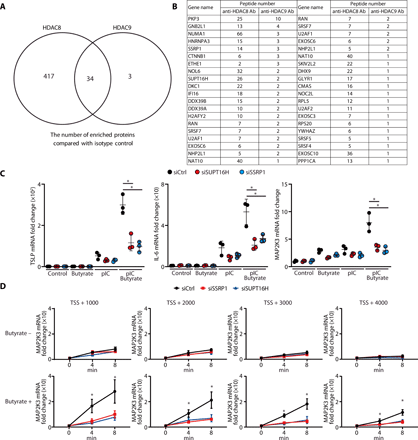
(A) Proteins identified by mass spectrometry of nHEK extracts enriched by immunoprecipitation with anti-HDAC8 or anti-HDAC9 compared with control IgG. (B) The common enriched nucleosome proteins precipitated by anti-HDAC8 and anti-HDAC9. (C) Transcriptional elongation FACT proteins SSRP1 and SUPT16H that were identified in (B) were silenced in nHEKs, and then HDAC activity was chemically inhibited by butyrate treatment (2 mM) for 1 hour. nHEKs were subsequently cultured with or without poly I:C (1 μg/ml) for 4 hours, and gene expression was measured by qPCR (N = 3; one-way ANOVA). (D) MAP2K3 transcriptional elongation assay. The transcriptional elongation rate of MAP2K3 was measured after SSRP1 or SUPT16H silencing with or without butyrate treatment (2 mM) for 1 hour. Data show the relative abundance of pulse-labeled RNA for the indicated genes at the indicated times after removal of the transcriptional inhibitor DRB (N = 3; one-way ANOVA). The results are expressed as the means ± SD. *P < 0.05. Data are representative of three independent experiments.
To our knowledge, a role for the FACT complex in regulation of inflammatory cytokine production in keratinocytes had not been previously reported. Therefore, to determine whether the association we observed with HDAC8/9 could be relevant to the effects we had observed on innate immune tolerance, we examined keratinocyte inflammatory responses after silencing of SSRP1 or SUPT16H. Keratinocytes were stimulated with poly I:C, and HDAC activity was inhibited with butyrate, a naturally occurring SCFA and pan-HDAC inhibitor in the skin that is produced by microorganisms through fermentation (13). Silencing of SSRP1 and SUPT16H blocked the capacity of butyrate to enhance TSLP, IL-6 (interleukin-6), and MAP2K3 expression in normal human keratinocytes (Fig. 2C and fig. S1, D and E). To directly measure the impact of the FACT complex on MAP2K3 gene elongation, transcriptional elongation assays were performed. Butyrate treatment increased MAP2K3 gene expression and promoted transcriptional elongation, and these functions were inhibited by SSRP1 and SUPT16H silencing (Fig. 2D). These results demonstrated how HDAC activity can act through the FACT complex to regulate transcription of MAP2K3.
MAP2K3 is directly influenced by HDAC8 and HDAC9 and is necessary for inhibition of cytokine expression by HDAC
To test whether HDAC8 or HDAC9 associates with MAP2K3, primary human keratinocytes were subjected to chromatin immunoprecipitation (ChIP) using anti-HDAC8 and anti-HDAC9 antibodies and then evaluated by ChIP-qPCR. Compared with the isotype control, a significant peak was observed within the MAP2K3 gene after immunoprecipitation with either anti-HDAC8 or anti-HDAC9 (Fig. 3A). Furthermore, silencing of HDAC8 and HDAC9 in human keratinocytes resulted in an increase in histone acetylation in MAP2K3 as assessed by ChIP-qPCR using anti-H3K9ac and anti-H3K27ac (Fig. 3B). These data show how both HDAC8 and HDAC9 can associate with the gene for MAP2K3, and acetylation of H3K9 and H3K27 marks within the MAP2K3 promoter will increase when the deacetylation activity from HDAC8 and HDAC9 is decreased (i.e., acetylation is increased) by inhibition of these HDACs.
(A) ChIP-qPCR for sites within the MAP2K3 gene relative to the transcriptional start site (TSS) after precipitation with anti-HDAC8 or anti-HDAC9 (N = 3; one-way ANOVA). (B) ChIP-qPCR for MAP2K3 gene relative to the TSS after precipitation with anti-H3K9ac or anti-H3K27ac. Data are shown in nHEKs after silencing of HDAC8, HDAC9, or control siRNA (N = 3; one-way ANOVA). (C) TNF-α, TSLP, or IL-6 mRNA expression in nHEKs measured by qPCR 4 hours after poly I:C stimulation and after silencing of MAP2K3 or control siRNA with or without pretreatment with butyrate (2 mM) for 1 hour (N = 3; Student’s t test). The results are expressed as the means ± SD. *P < 0.05. Data are representative of three independent experiments.
” data-icon-position=”” data-hide-link-title=”0″>
(A) ChIP-qPCR for sites within the MAP2K3 gene relative to the transcriptional start site (TSS) after precipitation with anti-HDAC8 or anti-HDAC9 (N = 3; one-way ANOVA). (B) ChIP-qPCR for MAP2K3 gene relative to the TSS after precipitation with anti-H3K9ac or anti-H3K27ac. Data are shown in nHEKs after silencing of HDAC8, HDAC9, or control siRNA (N = 3; one-way ANOVA). (C) TNF-α, TSLP, or IL-6 mRNA expression in nHEKs measured by qPCR 4 hours after poly I:C stimulation and after silencing of MAP2K3 or control siRNA with or without pretreatment with butyrate (2 mM) for 1 hour (N = 3; Student’s t test). The results are expressed as the means ± SD. *P < 0.05. Data are representative of three independent experiments.
To confirm the requirement for MAP2K3 to enable HDAC8 or HDAC9 to influence cytokine responses, we next examined the effect of silencing of MAP2K3 in keratinocytes. Control keratinocytes showed increased inflammatory cytokine expression [tumor necrosis factor–α (TNF-α), TSLP, and IL-6] when cells were activated with poly I:C, and HDAC activity was inhibited by the addition of butyrate, but the increase induced by HDAC inhibition would be largely lost if MAP2K3 was silenced (Fig. 3C and fig. S1F). This observation supports the conclusion that MAP2K3 mediates the increase in inflammatory cytokine expression after inhibition of HDAC activity. Together with our findings that specific HDAC8 and HDAC9 silencing recapitulates the effects of butyrate and that HDAC8 and HDAC9 associate with and alter histone acetylation of MAP2K3, our findings show that HDAC8 and HDAC9 confer tolerance to TLR stimuli by inhibition of MAP2K3 expression.
Expression of HDAC8/9 influences inflammatory responses of the skin
MAP2K3 has been proposed to be involved in inflammatory signaling in pulmonary epithelial cells through its action on p38MAPK (24–27). However, a role for regulation of MAP2K3 to control cytokine production by the skin was unknown. Analysis of expression of MAP2K3 in public microarray datasets from human inflammatory skin conditions (28–32) revealed that the expression of MAP2K3 was significantly up-regulated in skin exposed to ultraviolet (UV) radiation and in lesional skin of patients with atopic dermatitis, psoriasis, and acne vulgaris if compared with healthy skin (Fig. 4A). This observation in human clinical tissue supported the physiological relevance of MAP2K3 to skin inflammation and inspired us to further determine whether HDAC8 or HDAC9 directly influences MAP2K3 expression in the skin.
(A) MAP2K3 expression in human inflammatory skin disorders. Bar plots of z scores (y axis) for skin taken from healthy human controls and lesional skin taken from patients with UV radiation, psoriasis, atomic dermatitis (AD), and acne vulgaris were analyzed. Data were obtained from a public dataset (GEO accession numbers GDS2381, GSE13355, GDS968, and GDS2478) (Student’s t test, two-sided). (B) Ifnb1 and Tslp gene expression in the skin of K14-Cre Hdac8fl/fl and K14-Cre Hdac9fl/fl mice or controls as measured by qPCR 24 hours after UVB exposure (200 mJ/cm2) (N = 3; one-way ANOVA). (C) Ifnb1 gene expression in the skin of controls, K14-Cre Hdac9fl/fl, and double knockout K14-Cre Hdac8/9fl/fl 24 hours after UV radiation (N = 3; one-way ANOVA). (D) Ifnb1 gene expression (N = 3; one-way ANOVA) and ear swelling response after 7 days of topical IMQ application to controls, K14-Cre Hdac9fl/fl, and double knockout K14-Cre Hdac8/9fl/fl mice (controls, N = 3; K14-Cre Hdac9fl/fl mice, N = 3; double knockout K14-Cre Hdac8/9fl/fl mice, N = 4; one-way ANOVA). (E) Flow cytometry analysis of CD86 expression in Langerhans cells (LCs) isolated from the skin 24 hours after UVB exposure or after 7 days of IMQ application. Representative flow cytometry plots are shown on the left, and the MFI of CD86 is shown on the right [UV radiation experiment (N = 3), one-way ANOVA; IMQ application experiment (N = 4), Student’s t test]. (F and G) Flow cytometry analysis of cells gated from CD45+TCRβ1+ cells in the skin 24 hours after UVB exposure. Representative flow cytometry plots are shown as the number of CD4+ or CD8+ T cells in the skin is shown on the right (N = 3; Student’s t test). (H and I) Flow cytometry analysis of CD45+TCRβ1+ cells in the skin 24 hours after the final IMQ application on day 7. Representative flow cytometry plots are shown in (H), and the number of CD4+ or CD8+ cells in the skin is shown in (I) (N = 4; Student’s t test). The results are expressed as the means ± SD. Each dot represents an individual mouse. *P < 0.05. Data are representative of three independent experiments. WT, wild type.
” data-icon-position=”” data-hide-link-title=”0″>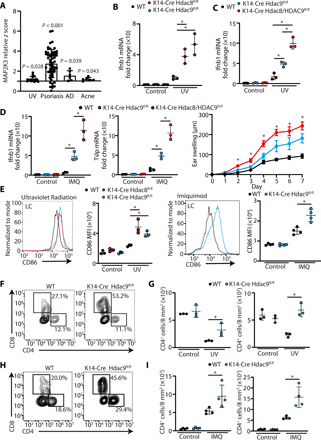
(A) MAP2K3 expression in human inflammatory skin disorders. Bar plots of z scores (y axis) for skin taken from healthy human controls and lesional skin taken from patients with UV radiation, psoriasis, atomic dermatitis (AD), and acne vulgaris were analyzed. Data were obtained from a public dataset (GEO accession numbers GDS2381, GSE13355, GDS968, and GDS2478) (Student’s t test, two-sided). (B) Ifnb1 and Tslp gene expression in the skin of K14-Cre Hdac8fl/fl and K14-Cre Hdac9fl/fl mice or controls as measured by qPCR 24 hours after UVB exposure (200 mJ/cm2) (N = 3; one-way ANOVA). (C) Ifnb1 gene expression in the skin of controls, K14-Cre Hdac9fl/fl, and double knockout K14-Cre Hdac8/9fl/fl 24 hours after UV radiation (N = 3; one-way ANOVA). (D) Ifnb1 gene expression (N = 3; one-way ANOVA) and ear swelling response after 7 days of topical IMQ application to controls, K14-Cre Hdac9fl/fl, and double knockout K14-Cre Hdac8/9fl/fl mice (controls, N = 3; K14-Cre Hdac9fl/fl mice, N = 3; double knockout K14-Cre Hdac8/9fl/fl mice, N = 4; one-way ANOVA). (E) Flow cytometry analysis of CD86 expression in Langerhans cells (LCs) isolated from the skin 24 hours after UVB exposure or after 7 days of IMQ application. Representative flow cytometry plots are shown on the left, and the MFI of CD86 is shown on the right [UV radiation experiment (N = 3), one-way ANOVA; IMQ application experiment (N = 4), Student’s t test]. (F and G) Flow cytometry analysis of cells gated from CD45+TCRβ1+ cells in the skin 24 hours after UVB exposure. Representative flow cytometry plots are shown as the number of CD4+ or CD8+ T cells in the skin is shown on the right (N = 3; Student’s t test). (H and I) Flow cytometry analysis of CD45+TCRβ1+ cells in the skin 24 hours after the final IMQ application on day 7. Representative flow cytometry plots are shown in (H), and the number of CD4+ or CD8+ cells in the skin is shown in (I) (N = 4; Student’s t test). The results are expressed as the means ± SD. Each dot represents an individual mouse. *P < 0.05. Data are representative of three independent experiments. WT, wild type.
To test the in vivo consequences of loss of HDAC8 and HDAC9 on skin inflammation, we generated mice with skin-specific loss of HDAC8 or HDAC9 or both by breeding Hdac8fl/fl and Hdac9fl/fl with mice with a skin-specific keratin 14 Cre driver (33, 34). To test the responsiveness to activation of TLR3, mice were exposed to UVB radiation (200 mJ/cm2). UV radiation was chosen as the initial model because this form of nonionizing radiation results in release of noncoding double-stranded RNA that is recognized by TLR3 (35), and the initial inflammatory signal is focused on surface epithelial keratinocytes. To test responsiveness to TLR7, imiquimod (IMQ) was applied to the skin of the ear for 7 days. Under conventional specific pathogen–free housing conditions, we did not observe a significant difference in interferon-β (IFN-β), TSLP, TNF-α, or CCL20 expression between control mice and mice lacking HDAC8 and HDAC9 in the skin (Fig. 4, B to D, and fig. S3, A and B). However, after UV radiation or IMQ application, inflammatory cytokine gene expression was significantly increased in mice without HDAC8 and HDAC9 in keratinocytes (Fig. 4, B to D, and fig. S3, A and B). Mice lacking both HDAC8 and HDAC9 had a greater increase in expression than mice with loss of HDAC9 alone (Fig. 4, C and D). These data suggested that tolerance to inflammation by UV damage or IMQ was lost in the absence of HDAC8 and HDAC9.
To further define the cell inflammatory responses in vivo to loss of HDAC8 or HDAC9, dendritic cell (DC) activation in the skin was assessed by fluorescence-activated cell sorting (FACS) analysis of CD86. No change in Langerhans cells, Langerin+ dermal DCs, and Langerin− dermal DCs was observed in the absence of an inflammatory challenge (Fig. 4E and fig. S3, C and D). However, consistent with prior observations of inflammatory cytokine responses and tissue inflammation, UV or IMQ exposure of K14-Cre Hdac9fl/fl and K14-Cre Hdac8fl/fl mice showed an increase in each DC subset compared with wild-type mice (Fig. 4E and figs. S3, C to F, and S4).
We next examined the T cell response in mouse skin lacking HDAC9. In normal skin, UV radiation results in an expected decrease in T cells (36, 37); however, the number of CD4+ and CD8+ cells increased in K14-Cre Hdac9fl/fl after UV radiation (Fig. 4, F and G). Loss of HDAC9 in the skin also resulted in an increase in CD4+ and CD8+ cells in mice exposed to topical IMQ (Fig. 4, H and I). Together, these observations show that the expression of HDAC8/9 in keratinocytes decreased the inflammatory response of the skin to UV radiation (triggered in part by TLR3) and IMQ, a TLR7-specific chemical ligand.
We next sought to determine whether the specific activity of HDAC8/9 influences responses to challenge by an infectious pathogen that can be detected by TLR2. To do this, we examined inflammation in response to Staphylococcus aureus (Fig. 5). Similar to the responses to UV and IMQ, topical application of S. aureus to K14-Cre Hdac9fl/fl mice resulted in increased cytokine expression (Fig. 5A). Topical application of HDAC8 or HDAC9 selective inhibitors also increased IFN-β expression by the skin (Fig. 5B). In addition, K14-Cre Hdac9fl/fl mice showed evidence of increased DC activation (Fig. 5C) and accumulation of CD4 and CD8+ T cells (Fig. 5D) compared with wild-type mice. Together, observations from three distinct mouse models of inflammation confirmed a role for HDAC8 and HDAC9 to suppress cutaneous inflammation and promote tolerance to a variety of inflammatory stimuli.
(A) Inflammatory gene expression in the skin measured by qPCR 24 hours after topical S. aureus application (N = 3; Student’s t test). (B) IFN-β gene expression in PCI34051- or TMP269-treated skin by qPCR 24 hours after topical S. aureus application (N = 3; one-way ANOVA). (C) Flow cytometry analysis of CD86 expression in the skin 24 hours after topical S. aureus application. Representative flow cytometry plots are shown on the left, and the MFI of CD86 is shown on the right (N = 3; Student’s t test). (D) Flow cytometry analysis of CD45+TCRβ1+ cells in the skin 24 hours after topical S. aureus application. Representative flow cytometry plots are shown on the left, and the number of CD4+ or CD8+ cells in the skin is shown on the right (N = 3; Student’s t test). The results are expressed as the means ± SD. Each dot represents an individual mouse. *P < 0.05. Data are representative of three independent experiments.
” data-icon-position=”” data-hide-link-title=”0″>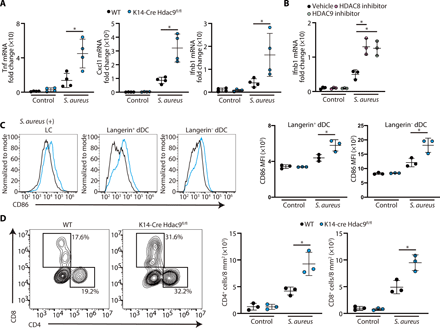
(A) Inflammatory gene expression in the skin measured by qPCR 24 hours after topical S. aureus application (N = 3; Student’s t test). (B) IFN-β gene expression in PCI34051- or TMP269-treated skin by qPCR 24 hours after topical S. aureus application (N = 3; one-way ANOVA). (C) Flow cytometry analysis of CD86 expression in the skin 24 hours after topical S. aureus application. Representative flow cytometry plots are shown on the left, and the MFI of CD86 is shown on the right (N = 3; Student’s t test). (D) Flow cytometry analysis of CD45+TCRβ1+ cells in the skin 24 hours after topical S. aureus application. Representative flow cytometry plots are shown on the left, and the number of CD4+ or CD8+ cells in the skin is shown on the right (N = 3; Student’s t test). The results are expressed as the means ± SD. Each dot represents an individual mouse. *P < 0.05. Data are representative of three independent experiments.
IFN-β is responsible for DC activation by keratinocytes after HDAC8 and HDAC9 silencing
To further clarify how the activity of HDAC8 or HDAC9 in keratinocytes can exert an effect on immune cells, human monocyte-derived DCs (MoDCs) were generated from peripheral blood then cultured with the condition media (CM) obtained from keratinocytes after silencing of HDAC8 or HDAC9 (Fig. 6A). CD86 expression was increased on MoDCs after exposure to CM from keratinocytes after silencing of HDAC8 or HDAC9 (Fig. 6, B and C). Addition of a neutralizing antibody to IFN-β abrogated this effect of keratinocyte CM (KC-CM) on MoDCs (Fig. 6C). This finding is consistent with observations that IFN-β is one of the factors derived from keratinocytes responsible for activation of DCs (8). Furthermore, we tested whether these activated DCs could influence T cell proliferation (Fig. 6D). An increase in T cell proliferation was observed in cells exposed to MoDC CM after activation by the CM of keratinocytes lacking HDAC8 or HDAC9 (Fig. 6E). Consistent with the earlier finding that neutralization of IFN-β could decrease DC activation, gene expression of IFN-β was significantly increased after silencing or application of inhibitors of HDAC8 and HDAC9 (Fig. 6, F and G). However, although CD86 expression on DC was significantly increased by CM derived from keratinocytes after inhibition of HDAC activity, this effect was canceled after silencing of MAP2K3 in keratinocytes (Fig. 6, H and I). Furthermore, IFNB1 gene expression was significantly suppressed by MAP2K3 silencing (Fig. 6J). These findings provide a new model to explain how keratinocytes regulate immune responses by the action of HDAC8 and HDAC9 on MAP2K3 activity and subsequent production of IFN-β (Fig. 7).
(A) Schematic showing the experimental approach; nHEKs (KC) were activated by poly I:C, and then KC-CM was transferred to human MoDCs for analysis by FACS. (B) Representative FACS histogram of CD86 expression in human MoDCs 24 hours after stimulation with KC-CM from HDAC8- or HDAC9-silenced nHEKs. (C) MFI from experiments shown in (B) (N = 3; one-way ANOVA). (D) Schematic showing the experimental approach as in (A) with transfer of DC to CD3+ cells for T cell proliferation assay. (E) The proliferation of CFSE-labeled CD3+ cells as analyzed by flow cytometry. (F and G) IFNB1 gene expression in nHEKs measured by qPCR after silencing of HDAC8/9 in (F) or treatment with PCI34051 (10 μM) or TMP269 (1 μM) in (G), followed by culture with or without poly I:C (1 μg/ml) (N = 3; one-way ANOVA). (H and I) Flow cytometry analysis of CD86+ human MoDCs stimulated with CM from nHEK after silencing of MAP2K3. nHEK CM was cocultured with peripheral blood CD3+ cells for 5 days. Representative flow cytometry histogram is shown in (H), and the MFI is shown in (I) (N = 3; Student’s t test). (J) IFNB1 mRNA expression in nHEK measured by qPCR after MAP2K3 silencing and treatment with poly I:C for 4 hours and after butyrate treatment (2 mM) (N = 3; Student’s t test). The results are expressed as the means ± SD. *P < 0.05. Data are representative of three independent experiments.
” data-icon-position=”” data-hide-link-title=”0″>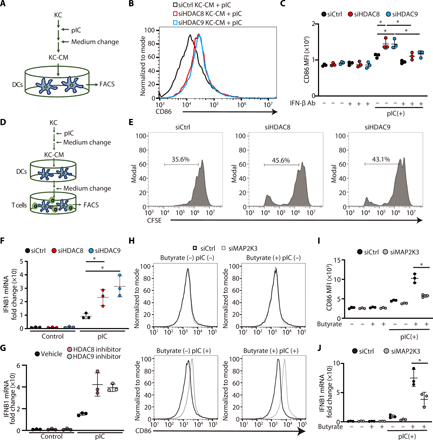
(A) Schematic showing the experimental approach; nHEKs (KC) were activated by poly I:C, and then KC-CM was transferred to human MoDCs for analysis by FACS. (B) Representative FACS histogram of CD86 expression in human MoDCs 24 hours after stimulation with KC-CM from HDAC8- or HDAC9-silenced nHEKs. (C) MFI from experiments shown in (B) (N = 3; one-way ANOVA). (D) Schematic showing the experimental approach as in (A) with transfer of DC to CD3+ cells for T cell proliferation assay. (E) The proliferation of CFSE-labeled CD3+ cells as analyzed by flow cytometry. (F and G) IFNB1 gene expression in nHEKs measured by qPCR after silencing of HDAC8/9 in (F) or treatment with PCI34051 (10 μM) or TMP269 (1 μM) in (G), followed by culture with or without poly I:C (1 μg/ml) (N = 3; one-way ANOVA). (H and I) Flow cytometry analysis of CD86+ human MoDCs stimulated with CM from nHEK after silencing of MAP2K3. nHEK CM was cocultured with peripheral blood CD3+ cells for 5 days. Representative flow cytometry histogram is shown in (H), and the MFI is shown in (I) (N = 3; Student’s t test). (J) IFNB1 mRNA expression in nHEK measured by qPCR after MAP2K3 silencing and treatment with poly I:C for 4 hours and after butyrate treatment (2 mM) (N = 3; Student’s t test). The results are expressed as the means ± SD. *P < 0.05. Data are representative of three independent experiments.
HDAC8 and HDAC9 are expressed in keratinocytes and decrease cytokine expression in response to commonly encountered inflammatory stimuli. When these HDACs are inhibited, keratinocytes mount a greater inflammatory response to stimulation by TLR ligands. This response includes the release of IFN-β that enables DC maturation and subsequent T cell proliferation. HDAC8 and HDAC9 act to decrease acetylation of the MAP2K3 gene. Inhibition of HDAC8 and HDAC9 leads to increased acetylation, activation of the FACT complex, and subsequent enhanced transcription of MAP2K3. This primes keratinocytes to increase cytokine expression in response to TLR stimuli.
” data-icon-position=”” data-hide-link-title=”0″>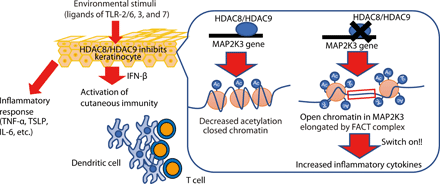
HDAC8 and HDAC9 are expressed in keratinocytes and decrease cytokine expression in response to commonly encountered inflammatory stimuli. When these HDACs are inhibited, keratinocytes mount a greater inflammatory response to stimulation by TLR ligands. This response includes the release of IFN-β that enables DC maturation and subsequent T cell proliferation. HDAC8 and HDAC9 act to decrease acetylation of the MAP2K3 gene. Inhibition of HDAC8 and HDAC9 leads to increased acetylation, activation of the FACT complex, and subsequent enhanced transcription of MAP2K3. This primes keratinocytes to increase cytokine expression in response to TLR stimuli.
DISCUSSION
Our study provides new insight into how the skin surface tolerates constant exposure to environmental and microbial compounds that would otherwise be expected to initiate a brisk inflammatory response. Inappropriate inflammatory responses of the epidermis are a hallmark of several human diseases, yet the factors that distinguish keratinocyte innate immune responses from classical immunocytes such as resident macrophages and DCs are poorly understood. Here, we show that transcriptional expression of MAP2K3 is a target for suppression by the action of HDAC8 and HDAC9. Our observations also illustrate how the FACT proteins SSRP1 and SUPT16H are critical for the expression of MAP2K3 in keratinocytes, thus defining additional checkpoints in this system. Furthermore, we show that HDAC8- and HDAC9-deficient keratinocytes produce increased IFN-β that then activates DC function to subsequently lead to T cell proliferation. These observations provide new insight into the maintenance of innate immune tolerance.
Several independent lines of evidence support the conclusion that MAP2K3 is directly affected by HDAC8 and HDAC9, thus providing epigenetic control of keratinocyte immune function. There are few reported studies focused on MAP2K3-related skin diseases (38). Our analysis of previously published transcriptional data from human skin diseases showed that expression of MAP2K3 increases in major inflammatory skin diseases such as atopic dermatitis, psoriasis, and acne vulgaris. This association is consistent with our conclusions that regulation of MAP2K3 function may be a key checkpoint for maintaining homeostasis. Furthermore, additional studies have shown that inhibition of the MAP2K3 downstream target p38MAPK impairs inflammatory responses in IMQ-induced psoriasis inflammation (39), C. acnes–induced skin inflammation (40), and UV radiation–induced skin inflammation (41). Thus, both MAP2K3 and p38MAPK inhibition are potential therapeutic strategies for the control of unwanted skin inflammation.
A putative binding site for HDAC8 and HDAC9 was identified in a location distant from the promoter region of MAP2K3 gene and may represent an enhancer location. However, both H3K9ac and H3K27ac marks in the promoter region of MAP2K3 were increased after silencing of HDAC8 and HDAC9. Therefore, we believe that both HDAC8 and HDAC9 can influence acetylation in the promoter region of MAP2K3. We speculate that a DNA loop structure may be involved that would lead to contact between the HDAC8/9 binding region and the MAP2K3 promoter region (42). Although we could not identify its mechanism in this study, further analysis of the structure of the MAP2K3 gene is warranted.
It remains unclear what the natural impact is of HDAC inhibition by SCFAs derived from skin commensal bacteria. Under normal physiological conditions, SCFAs produced in the skin should easily penetrate into the dermis because of their low molecular weight of less than 500 Da (43). The topical application of butyrate impairs acquired immune responses in the skin (44). This observation is consistent with the opposing effects that inhibition of HDACs has on bone marrow–derived immunocytes such as macrophages (13). Unlike keratinocytes that become less tolerant of TLR stimuli upon inhibition of HDAC, macrophage responses are inhibited due to increased expression of repressor genes. A complex contextual immune regulatory system within the three-dimensional structure of the skin must tolerate danger signals at the surface and appropriately respond to danger. In the example of the skin disease acne, it has been assumed that occluded hair follicles under fermentation conditions metabolize lipid-enriched materials secreted from sebaceous glands into SCFAs. Because the lipid-enriched molecules are able to be retained in the epidermis or stratum corneum (45), SCFAs in hair follicles at sufficient concentrations can then exert cell-dominant effects on the keratinocytes rather than the dermal cells. From the perspective of microbial ecology, production of SCFAs might have some advantages for commensal bacteria to survive at the expense of other bacteria if an increased host inflammatory response selectively targets the competition.
The different inflammatory responses between epithelial cells and bone marrow cells illustrate how epigenetic modifications are cell type dependent and can have opposite effects. In macrophages, the presence of the corepressor and the nucleosome remodeling and deacetylase complex (NuRD) suppresses expression of inflammatory cytokines (46, 47). Inhibition of HDAC activity will increase NuRD expression and therefore suppress expression of inflammatory cytokines. In contrast, this complex is not active in keratinocytes (13), and inhibition of HDAC8 and HDAC9 positively drives inflammation, a response that is opposite from macrophages. Therefore, similar epigenetic changes can have either a proinflammatory or anti-inflammatory response depending on the cell type that is targeted.
The FACT complex is involved in transcription-related chromatin dynamics (48), promotes transcriptional elongation, and contributes to the development of liver cancer (49). Our observations in keratinocytes suggest that HDAC8/9 and the FACT complex are powerful genetic regulators that have opposite effects. This may enable rapid acceleration of inflammatory responses upon exposure to SCFAs. Prior reports have suggested that inhibition of the FACT complex is beneficial in an atopic dermatitis model (50), an IMQ-induced psoriasis model (51), and after UV radiation (52). A clinical trial in patients with psoriasis of oral administration of a FACT complex inhibitor was not therapeutically successful (53). More work is needed to understand the role of components of the FACT complex, but our findings suggest that local topical application of agents targeting the pathways defined in this study could be useful for the treatment of inflammatory skin diseases.
MATERIALS AND METHODS
Study design
The aim of this study was to investigate the role of HDAC8 and HDAC9 in skin inflammation. We performed inhibited expression of HDAC8 or HDAC9 in keratinocytes and evaluated inflammatory responses after exposure to TLR2/6, TLR3, or TLR7 ligands. ChIP-seq and signal pathway analysis by RNA-seq, as well as antibody pulldown and mass spectrometry, were used to identify the targets of HDAC8 and HDAC9. Mice deficient in HDAC8 and/or HDAC9 were used to assess the in vivo contribution of HDAC8/9 to inflammatory triggers including UV radiation, IMQ, and topical S. aureus. Keratinocytes were assessed in vitro to determine how loss of HDAC8/9 activity can activate DCs and subsequent T cell proliferation.
Animals and animal care
All experiments involving mice were done with the approval of the Institutional Animal Care and Use Guidelines of the University of California, San Diego (protocol number S09074). Eight- to 12-week-old mice were used in this study. All animals were in the C57BL/6 genetic background. K14-Cre mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Hdac8fl/fl mice were purchased from Taconic Biosciences (Hudson, NY) under a material transfer agreement. Hdac9fl/fl mice were provided from Augusta University and originally generated by N. L. Weintraub. To prepare keratinocyte-specific Hdac8- and Hdac9-deficient mice, mice bearing the Cre recombinase transgene, under the control of the keratin 14 promoter (K14-Cre), were mated with Hdac8fl/fl or Hdac9fl/fl mice.
Mouse inflammation induced by UV radiation, IMQ application, or S. aureus
UV radiation was performed as previously described with some modification (54). Briefly, 92 hours after the treatment of hair shave (Wahl, Sterling, IL) and chemical hair remover (Church & Dwight Co. Inc., Princeton, NJ), UV (200 mJ/cm2) was radiated on the back skin by a UV radiator (Spectroline model EB-280C UV lamp; Spectronics Corp., Westbury, NY). The total amount of UV was measured by a solar meter (Solartech Inc., Harrison Township, MI). The skin sample was collected for further experiments. IMQ (10 mg) was daily applied on the ear skin for six consecutive days, and the ear thickness was measured by a thickness gauge (Teclock, Japan). The skin sample was collected on day 7 for further experiments.
After shaving of the back skin and application of a chemical depilatory agent, S. aureus USA300 (106 colony-forming units) was placed on 8-mm2 tryptic soy broth (TSB) agar, which was applied on the back skin by the fixation of Tegaderm (3M Health Care, Germany). The skin sample was collected 24 hours after the treatment for further experiments.
Topical HDAC inhibitor treatment was conducted for 24 hours before S. aureus application. Agar plates (2%) were made by using UltraPure Agarose (Invitrogen) containing PCI34051 (1 mM) and TMP269 (100 μM). Eight-millimeter agar discs were made to include 10 μl of distilled H2O, PCI34051 (1 mM), or TMP269 (100 μM). These discs were then topically applied to the dorsal skin and fixed in place using Tegaderm (3M Company).
Keratinocyte culture
Normal human epidermal keratinocytes (nHEKs) (Thermo Fisher Scientific, Waltham, MA) were cultured with EpiLife medium supplemented with 60 mM CaCl2 (Thermo Fisher Scientific), 1× EpiLife Defined Growth Supplement (Thermo Fisher Scientific), and 1× antibiotic/antimycotic (Thermo Fisher Scientific).
Silencing HDAC8, HDAC9, MAP2K3, SSRP1, and SUPT16H by siRNA
HDAC8, HDAC9, and MAP2K3 in nHEKs were knocked down using Silencer Select small interfering RNA (siRNA) (Thermo Fisher Scientific) according to the manufacturer’s protocol with some modifications. Briefly, 100 nM siRNA (Thermo Fisher Scientific) in Opti-MEM (Thermo Fisher Scientific) with 2.5% Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific) was incubated at room temperature for 5 min and then diluted in 1:10 ratio in EpiLife culture medium supplemented with 1× EpiLife Defined Growth Supplement and 1× antibiotic/antimycotic to set a final concentration of siRNA and Lipofectamine as 10 nM and 0.25%, respectively. After siRNA treatment for 16 hours, siRNA-treated nHEKs were recovered for 48 to 72 hours before further experiments.
Quantitative real-time PCR
Total RNA was extracted from the skin using a TRIzol RNA extraction reagent (Invitrogen, Carlsbad, CA). Cultured cells were lysed, and RNA was isolated using the PureLink RNA Isolation Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Complementary DNA (cDNA) was reverse-transcribed from the total RNA samples using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Berkeley, CA). cDNA was used in quantitative real-time PCR using TaqMan Universal PCR Master Mix (Thermo Fisher Scientific) and TaqMan Gene Expression Assays (Thermo Fisher Scientific) or iTaq Universal SYBR Green Supermix (Bio-Rad). The gene expression was measured by a CFX96 Real-Time System (Bio-Rad) and was determined by ΔΔCt. The results were normalized to those of the housekeeping glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA.
Immunoblotting
nHEKs were lysed by radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific) with protease inhibitor (Sigma-Aldrich). After passing the lysate 20 times through a 21-gauge needle attached to a ribonuclease (RNase)–free syringe (BD, Franklin Lakes, NJ), lysates were sonicated in ice water for 20 min. After centrifuging for 20 min at 16,000 rpm, the supernatant was collected. Protein concentration was calculated by a bicinchoninic acid assay (Pierce Biotechnology, Rockford, IL). Samples were mixed with 4× Laemmli sample buffer (Bio-Rad) and were incubated at 95°C for 10 min. Proteins from the total cell lysate were separated by Mini-PROTEAN TGX precast gel (Bio-Rad) and transferred to the Trans-Blot Turbo mini polyvinylidene difluoride membrane (Bio-Rad) using Trans-Blot Turbo Instrument (Bio-Rad). The membrane was washed by Odyssey blocking solution (LI-COR Biosciences, Lincoln, NE) for 1 hour at room temperature and then stained with anti-MAP2K3 (Abcam, Cambridge, MA), anti–phospho-p38MAPK, anti-p38MAPK (Cell Signaling Technology, Danvers, MA), anti-HDAC8, or anti-HDAC9 antibodies (Abcam) and subsequently treated with secondary antibody (LI-COR Biosciences). GAPDH was used as an internal control and was stained by anti-GAPDH antibody (Fitzgerald Industries, Concord, MA). The membrane was washed, and the immunoblot signals were detected by the Odyssey infrared imaging system (LI-COR Biosciences).
Chromatin immunoprecipitation PCR
ChIP-qPCR was performed as previously described (13). Samples were cross-linked in 1% formaldehyde in phosphate-buffered saline (PBS) for 10 min at room temperature, and the reaction was quenched by 125 mM glycine for 5 min at room temperature. After washing with PBS twice, adhesive cells were scraped by a cell scraper (Thermo Fisher Scientific), and the sample pellet was collected into 10 ml of PBS in a conical tube by centrifuge at 600g for 5 min. The cell pellet was lysed by lysis buffer [5 mM Pipes (pH 8.0), 85 mM KCl, and 0.5% NP-40] with 1× protease inhibitor (Sigma-Aldrich) on ice for 5 min, and the pellet was collected by centrifuge at 400g for 5 min. After resuspension by RIPA buffer with 1× protease inhibitor (Sigma-Aldrich), the sample was sonicated by a Diagenode Bioruptor sonicator (4 × 7.5-min cycles of 30 s on and 60 s off) (Diagenode Inc., Sparta, NJ) and centrifuge at 16,000g for 15 min at 4°C. The supernatant was collected and then used for immunoprecipitation treatment. The sonicated sample was incubated with beads conjugated with anti-HDAC8, anti-HDAC9, anti-H3K9ac, or anti-H3K27ac antibodies (Abcam) or isotype control immunoglobulin G (IgG) at 4°C overnight. After washing with wash buffer [100 mM tris (pH 7.5), 500 mM LiCl, 1% NP-40, and 1% sodium deoxycholate] and once with tris-EDTA, beads were suspended with 200 μl of immunoprecipitation elution buffer (1% sodium deoxycholate and 0.1 M NaHCO3). The cross-linked chromatin was eluted from the beads by incubation at 65°C for 1 hour and centrifugation at 16,000g for 3 min. The supernatant was collected, and cross-link reversal treatment was performed by incubation at 65°C overnight. Immunoprecipitated DNA was purified using a Qiagen cleanup kit (Qiagen, Hilden, Germany), and samples were used for ChIP-seq or qPCR along with input DNA.
FACS analysis
To prepare single-cell suspensions, skin sample was taken from the back skin by using 8-mm2 disposable biopsy punches (Integra-Miltex, York, PA) and treated by collagenase D (2.5 mg/ml; Sigma-Aldrich) and deoxyribonuclease 1 (30 ng/ml; MP Biomedicals, Solon, OH) for 2 hours at 37°C. After filtration by using a 40-μm cell strainer (Thermo Fisher Scientific), skin suspension cells were stained by various combinations of fluorescence-conjugated monoclonal antibodies (mAbs) and were analyzed by ZE5 Cell Analyzer (Bio-Rad) and FlowJo software (TreeStar, San Carlos, CA). For cell analysis, the following antibodies were used: allophycocyanin (APC)–conjugated anti-CD11c and Langerin mAbs (BioLegend), APC-eFluor 780–conjugated anti-CD4 mAb (Invitrogen), BV-conjugated anti-CD45 mAb (BioLegend), BV680-conjugated anti-CD8 mAb (BioLegend), fluorescein isothiocyanate–conjugated anti-CD86 (BioLegend), HLA-DR (BioLegend), major histocompatibility complex class II (eBioscience, San Diego, CA) mAbs, phycoerythrin-conjugated anti-EpCAM and CD86 mAbs (eBioscience), Per-CP5.5–conjugated anti-CD11c (eBioscience), and TCRβ (Invitrogen) mAbs. The cell number was counted using CountBright Absolute Counting Beads (Thermo Fisher Scientific).
RNA-seq analysis
RNA extracts taken from nHEKs were submitted to the University of California, San Diego (UCSD) Institute for Genomic Medicine core facility for quality analysis, library preparation, and high-throughput sequencing. The RNA quality was assessed using the Agilent TapeStation instrument (Agilent Technologies, Santa Clara, CA). Libraries were constructed using TruSeq Stranded mRNA Library Prep Kits (Illumina, San Diego, CA) and were analyzed on a HiSeq 4000 instrument (Illumina). The raw data obtained from the UCSD Institute for Genomic Medicine core facility were analyzed using Partek Flow software (Partek Inc., St. Louis, MO) to determine transcript abundance and identify differentially expressed genes.
MoDC preparation
Monocytes were isolated from peripheral blood mononuclear cells obtained from healthy blood donors by the Ficoll-Paque method (Pharmacia, Uppsala, Sweden). CD14+ cells purified with MACS bead (Miltenyi Biotec, Auburn, CA) were cultured in complete RPMI (RPMI 1640; Sigma-Aldrich, St. Louis, MO) containing 10% heat-inactivated fetal calf serum (Gemini Bioproducts, Calabasas, CA), 50 μM 2-mercaptoethanol (Sigma-Aldrich), 2 mM l-glutamine (Corning), 25 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 1 mM nonessential amino acids (Life Technologies), 1 mM sodium pyruvate (GE Healthcare), penicillin (100 U/ml), and streptomycin (100 μg/ml; Life Technologies), supplemented with recombinant human granulocyte-macrophage colony-stimulating factor (100 ng/ml; R&D Systems, Minneapolis, MN) and recombinant human IL-4 (50 ng/ml; R&D Systems) for 7 days.
DC activation and T cell proliferation
MoDC was cocultured with the CM from nHEKs with poly I:C (Invitrogen) and/or a neutralizing anti–IFN-β antibody (BioLegend, San Diego, CA) for 24 hours, and then the degree of CD86 expression was counted by flow cytometry as mean fluorescence intensity (MFI). In addition, to measure the degree of T cell proliferation, autologous naïve CD3+ cells were sorted from human peripheral blood using MACS bead (Miltenyi Biotec) and then stained by CellTrace CFSE (carboxyfluorescein diacetate succinimidyl ester) (Thermo Fisher Scientific). After stimulation with KC-CM and/or IFN-β–neutralizing antibody for 24 hours, MoDCs were then washed to remove KC-CM before adding to CD3+ cells in a ratio of 1:5 (DCs: T cells) under KC-CM stimulation. Five days after the culture, the intensity of CFSE fluorescence was determined by flow cytometry.
Immunoprecipitation for mass spectrometry
nHEKs were washed with cold PBS and scraped from the plates using a cell scraper. After transferring the cells from the plates to a 15-ml conical tube, samples were centrifuged for 5 min at 500g at 4°C. After the aspiration of supernatant, cell pellets were frozen on dry ice and transferred to −80°C for storage until ready for cell lysis and affinity purification. Pellets were treated with lysis buffer [50 mM Hepes-NaOH (pH 8.0), 100 mM KCl, 2 mM EDTA, 0.1% NP-40, and 10% glycerol] with protease inhibitor (Sigma-Aldrich). After passing the lysate 20 times through a 21-gauge needle attached to an RNase-free syringe (BD, Franklin Lakes, NJ), lysates were subjected to two freeze-thaw cycle treatments by incubating the tube on dry ice for 10 min and then transferring it to a 37°C water bath. After centrifuging for 20 min at 16,000 rpm at 4°C, the supernatant was collected and used for immunoprecipitation treatment by incubation with bead-conjugated anti-HDAC8, anti-HDAC9 (Abcam), or isotype control IgG at 4°C overnight. After three washes with lysis buffer containing 50 mM ammonium bicarbonate, bead-conjugated proteins were used for mass spectrometry analysis.
4sUDRB elongation assay
First, the gene transcription was inhibited by 5,6-dichlorobenzimidazole 1-β-d-ribofuranoside (DRB) (Sigma-Aldrich) at a final concentration of 100 μM for 3 hours. Then, 4-thiouridine (Sigma-Aldrich) was pulsed at a final concentration of 1 mM to label newly transcribed RNA. Total RNA was extracted with the miRNeasy kit (Qiagen) 0, 4, and 8 min after DRB removal. 4sU-labeled RNA was biotinylated by EZ-Link Biotin-HPDP (Thermo Fisher Scientific) dissolved in dimethylformamide (Thermo Fisher Scientific) and purified by using Dynabeads MyOne Streptavidin T1 beads (Life Technologies). 4sU-labeled RNA was eluted with 100 mM dithiothreitol (Thermo Fisher Scientific). After cleaning up by using the RNeasy MinElute Kit (Qiagen), eluted RNA was used for qPCR amplifications by using specific primers.
Microarray data analysis
Expression data for patients with atopic dermatitis, psoriasis, allergic contact dermatitis, and UV light radiation were obtained from a public dataset deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database (GEO accession numbers GDS2381, GSE13355, GDS968, and GDS2478). Patient and sample information was previously described (29, 31, 32, 55).
Statistical analysis
All statistical analyses were calculated using GraphPad Prism 6.05 (GraphPad Prism Software Inc., La Jolla, CA). Student’s t test was used to calculate significant differences between the two groups. Statistical significance between multiple groups was analyzed using the analysis of variance (ANOVA) comparisons test. All P values less than 0.05 were considered statistically significant.
Acknowledgments: We appreciate N. L. Weintraub in Augusta University for providing Hdac9fl/fl mice. Funding: R.L.G. is supported by NIH grants R01AR069653, R01AR074302, R01AR076082, R37AI052453, and U01AI52038. Y.S. is supported by a JSPS overseas fellowship. Author contributions: Y.S. conducted all the experiments and wrote the manuscript; T.D. and M.C.L. conducted RNA-seq analysis; T.N. designed and supervised the topical S. aureus application experiment; N.N.K. designed the UV radiation experiment; G.S. supervised the epigenetic experiments; and R.L.G. supervised and organized all experiments and wrote the paper. Competing interests: R.L.G. is a cofounder, scientific advisor, and consultant and has equity in MatriSys Bioscience and is also a consultant, receives income, and has equity in Sente Inc. The other authors declare that they have no competing interests. Data and materials availability: RNA-seq data were deposited in the NCBI GEO database (GEO accession no. GSE154163). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.